
基因定點突變全攻略
信息來源:金開瑞 作者:genecreate 發布時間:2022-11-01 10:41:35
一、定點突變的目的
把目的基因上面的一個堿基換成另外一個堿基。
二、定點突變的原理
定點突變是指通過聚合酶鏈式反應(PCR)等方法向目的DNA片段(可以是基因組,也可以是質粒)中引入所需變化(通常是表征有利方向的變化),包括堿基的添加、刪除、點突變等。定點突變能迅速、高效的提高DNA所表達的目的蛋白的性狀及表征,是基因研究工作中一種非常有用的手段。
體外定點突變技術是研究蛋白質結構和功能之間的復雜關系的有力工具,也是實驗室中改造/優化基因常用的手段。蛋白質的結構決定其功能,二者之間的關系是蛋白質組研究的重點之一。對某個已知基因的特定堿基進行定點改變、缺失或者插入,可以改變對應的氨基酸序列和蛋白質結構,對突變基因的表達產物進行研究有助于人類了解蛋白質結構和功能的關系,探討蛋白質的結構/結構域。而利用定點突變技術改造基因:比如野生型的綠色熒光蛋白(wtGFP)是在紫外光激發下能夠發出微弱的綠色熒光,經過對其發光結構域的特定氨基酸定點改造,現在的GFP能在可見光的波長范圍被激發(吸收區紅移),而且發光強度比原來強上百倍,甚至還出現了黃色熒光蛋白,藍色熒光蛋白等等。定點突變技術的潛在應用領域很廣,比如研究蛋白質相互作用位點的結構、改造酶的不同活性或者動力學特性,改造啟動子或者DNA作用元件,提高蛋白的抗原性或者是穩定性、活性、研究蛋白的晶體結構,以及藥物研發、基因治療等等方面。
通過設計引物,并利用PCR將模板擴增出來,然后去掉模板,剩下來的就是我們的PCR產物,在PCR產物上就已經把這個點變過來了,然后再轉化,篩選陽性克隆,再測序確定就行了。
三、引物設計原則
引物設計的一般原則不再重復。
突變引物設計的特殊原則:
(1)通常引物長度為25~45 bp,我們建議引物長度為30~35 bp。一般都是以要突變的堿基為中心,加上兩邊的一段序列,兩邊長度至少為11-12 bp。若兩邊引物太短了,很可能會造成突變實驗失敗,因為引物至少要11-12個bp才能與模板搭上,而這種突變PCR要求兩邊都能與引物搭上,所以兩邊最好各設至少12個bp,并且合成多一條反向互補的引物。
(2)如果設定的引物長度為30 bp,接下來需要計算引物的Tm值,看是否達到78℃(GC含量應大于40%)。
(3)如果Tm值低于78℃,則適當改變引物的長度以使其Tm值達到78℃(GC含量應大于40%)。
(4)設計上下游引物時確保突變點在引物的中央位置。
(5)最好使用經過純化的引物。
Tm值計算公式:Tm=0.41×(% of GC)–675/L+81.5
注:L:引物堿基數;% of GC:引物GC含量。
四、引物設計實例
以GCG→ACG為例:
5’-CCTCCTTCAGTATGTAGGCGACTTACTTATTGCGG-3’
(1)首先設計30 bp長的上下游引物,并將A (T)設計在引物的中央位置。
Primer #1: 5’-CCTTCAGTATGTAGACGACTTACTTATTGC-3’
Primer #2: 5’-GCAATAAGTAAGTCGTCTACATACTGAAGG-3’
(2)引物GC含量為40%,L為30,將這兩個數值帶入Tm值計算公式,得到其Tm=75.5(Tm=0.41×40-675/30+81.5)。通過計算可以看出其Tm低于78℃,這樣的引物是不合適的,所以必須調整引物長度。
(3)重新調整引物長度。
Primer #1: 5’-CCTCCTTCAGTATGTAGACGACTTACTTATTGCGG-3’
Primer #2: 5’-CCGCAATAAGTAAGTCGTCTACATACTGAAGGAGG-3’
在引物兩端加5mer(斜體下劃線處),這樣引物的GC含量為45.7%,L值為35,將這兩個數值帶入Tm值計算公式,得到其Tm為80.952(Tm=0.41×47.5-675/35+81.5),這樣的引物就可以用于突變實驗了。
五、突變所用聚合酶及Buffer
引物和質粒都準備好后,當然就是做PCR嘍,不過對于PCR的酶和buffer,不能用平時的,我們做PCR把整個質粒擴出來,延伸長度達到幾個K,所以要用那些GC buffer或擴增長片段的buffer,另外,要用保真性能較好的PFU酶來擴增,防止引進新的突變。
除了使用基因定點突變試劑盒,如Stratagene和塞百盛的試劑盒,但價格昂貴。可以使用高保真的聚合酶,如博大泰克的金牌快速taq酶、Takara的PrimeSTARTM HS DNA polymerase。
六、如何去掉PCR產物
最簡單的方法就是用DpnI酶,DpnI能夠識別甲基化位點并將其酶切,我們用的模板一般都是雙鏈超螺旋質粒,從大腸桿菌里提出來的質粒一般都被甲基化保護起來(除非你用的是甲基化缺陷型的菌株),而PCR產物都是沒有甲基化的,所以DpnI酶能夠特異性地切割模板(質粒)而不會影響PCR產物,從而去掉模板留下PCR產物,所以提質粒時那些菌株一定不能是甲基化缺陷株。
DpnI處理的時間最好長一點,最少一個小時吧,最好能有兩三個小時,因為如果模板處理得不干凈,哪怕只有那么一點點,模板直接在平板上長出來,就會導致實驗失敗。
七、如何拿到質粒
直接把通過DpnI處理的PCR產物拿去做轉化就行了,然后再篩選出陽性克隆,并提出質粒,拿去測序,驗證突變結果。
八、圖示
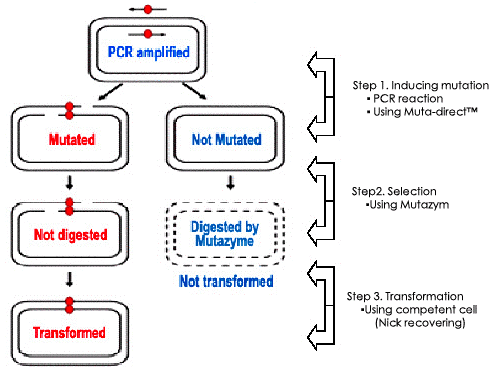
九、定點突變操作步驟
[A] 誘導突變基因(PCR反應)以待突變的質粒為模板,用設計的引物及Muta-direct™酶進行PCR擴增反應,誘導目的基因突變。
1. 設計點突變引物。
[注]參考引物設計指導
2. 準備模板質粒DN A
[注]用dam+型菌株(例如DH5α菌株)作為宿主菌。在end+型菌株中常有克隆數低的現象,但是對突變效率沒有影響。提取質粒DNA時我們建議您使用本公司的質粒提純試劑盒。
3. [選項]對照反應體系(50μl反應體系)
4. 樣品反應體系(50μl反應體系)
5. PCR反應條件
[注]按如下參數設置PCR擴增條件。
6. PCR擴增反應完成后冰育5分鐘,然后置于室溫(避免反復凍融)。
[注] 按下列提供的PCR條件進行擴增,控制PCR循環數。注意當突變點位點超過4個時會發生突變率降低的現象。
[B] 突變質粒選擇
PCR反應結束后使用Mutazyme™酶消化甲基化質粒從而選擇突變質粒DNA。
1. 準備PCR反應產物
2. 加入1μl(10U/μl)Mutazyme™酶37℃溫育1小時。
[注]當質粒DNA用量過多時Mutazyme™酶可能發生與樣品反應不完全的現象。因此我們建議為了保證突變率請嚴格遵照實驗步驟進行操作。如果突變率低,可以適當延長反應時間或增加Mutazyme™酶用量。
[C]轉化
反應完畢后在質粒DNA上會產生缺口,當把這個質粒DNA轉入E.coli中時請選擇dam+型菌株,例如DH5α。
1. 將10μl樣品加到50μl感受態細胞里,然后放置在冰上30分鐘。
2. 接下來可以參照一般的轉化步驟進行。
序列分析
通常當LB平板上出白色菌落則表明發生了突變。
為了證實這一結果,我們建議對白色單菌落進行測序分析。
先講最簡單的一個點的定點突變技術,其它較長片段的突變,刪除,插入技術以后會慢慢奉上:在做實驗之前,我們首先要搞清楚實驗的目的和實驗的原理。
實驗的目的應該比較明確吧:就是要把自己的基因上面的一個堿基換成另外一個堿基。一般情況下我們會有幾種可能使我們需要這樣去做:
第一:我們吊出來的基因有點突變,相信這可能是大家經常會遇到的問題。基因好不容易吊出來,并裝進了自己的載體,卻發現有一兩個堿基跟自己的預期序列或所有的公共數據庫不匹配,然后暴昏。
大家實驗室里面還是用Taq酶為主吧,Pfu這樣的高保真酶大家應該用得不多吧,Taq酶的優點和缺點都很明顯:優點就是擴增效能強,缺點就是保真性差,其錯配機率是比較高的,相關數字忘了,大家可以去網上查那個數字,不過感覺如果是2000bp的基因,如果擴四五十個循環的話,很大機率會出現點突變,當然這也跟具體PCR體系里的Buffer有很大關系,詳細情況這里就不討論了。
第二:要研究基因的功能,在基因上自己選定位置更換堿基的保守序列,或者改造成不同的亞型,總之就是要人工改造堿基序列符合自己的實驗需要,相信這也是那些研究基因的人經常的一種思路吧。
對于第一種情況:我們首先要分析出現堿基不匹配的位置是不是重要的位置,如果不是很重要,大可不必管它,比如說是三聯密碼子的最后一位,堿基的改變并沒有引起相應氨基酸的改變,那么一般情況下也可以不去理它。另外,在NCBI上人類的基因的版本一直在變化,也就是說同一個基因有不同的版本,或者稱不同的亞型,其堿基序列有些許的差異,只要自己克隆出來的堿基序列與其中一個相匹配,一般也就可以不做定點突變了。如果有時間沒錢,那干脆重新PCR然后再克隆進自己的載體了,不過最好換個保真性好一點的酶如PFU,或者PCR循環數低一點,不過這些東西有時候也得靠運氣啦。實在不行的話再來做定點突變。
對于第二種情況:這種情況下一般也就只能做定點突變了。
接下來開始聊一聊定點突變的原理吧,那個Stratagene試劑盒!上面有一個說明書,說得好像很正規,不過上面好多都是什么專利啊什么注意之類的話,看都不看,我們簡明扼要地只講實驗方面,通過設計引物,并利用PCR將模板擴增出來,然后去掉模板,剩下來的就是我們的PCR產物,在PCR產物上就已經把這個點變過來了,然后再轉化,篩選陽性克隆,再測序確定就行了。
大家馬上就會想到幾個問題了:
第一:引物怎么設計呢?
第二:模板怎么去掉呢?
第三:怎么拿到質粒呢?
對于第一個問題:怎么設計引物?
我只能講一些原則,并舉一些例子。
引物設計的原則其它貼子上都有講,這里就不重復了:
不過這種突變引物要加上一個原則:
一般都是以要突變的堿基為中心,加上兩邊的一段序列,兩邊長度至少為11-12base pair。
若兩邊引物太短了,很可能會造成突變實驗失敗,大家應該都知道,引物至少要11-12個base pair才能與模板搭上,而這種突變PCR要求兩邊都能與引物搭上,所以兩邊最好各設至少12個base pair,并且合成多一條反向互補的引物。
這么說大家可能不是很清楚,那我就舉個例子吧:
X71661.1 TATCAGGAGGAATTTGAGCACTTTCAACAAGAATTGGATAAAAAAAAAGAGGAATTCCAG 960
現有序列 TATCAGGAGGAATTTGAGCACTTTCAACAAGAATTGGATAAAAAAAAAGAGGAATTCCAG 924
*********************************************** ************
|----deletion
X71661.1 AAGGGCCACCCCGACCTCCAAGGGCAGCCTGCGGAGGAAATATTTGAGAGTGTAGGAGAT 1020
現有序列 AAGGGCCACCCCGACCTCCAAGGGCAGCCTGCGGAGGAAATATTTGAGAGTGTAGGAGAT 984(上面為目的序列,下面為現有序列:我們發現有一個A堿基的缺失,其直接結果是在表達蛋白時后面的氨基酸全部錯配)
我們以它為中心設計引物:兩邊各至少12個堿基,左邊由于含有較多的A造成引物GC%含量過低,故拉長引物使GC%含量不至過低,也使引物退火溫度升高。
故合成引物CAACAAGAATTGGATAAAAAAAAAGAGGAATTCCAGAAG
并合成反向互補引物CTTCTGGAATTCCTCTTTTTTTTTATCCAATTCTTGTTG
其實也不一定要反向互補序列,只要反向引物也是兩邊都有大于12個堿基,同時符合引物設計的原則就行了。
引物合成公司有很多家,大家可以去尋找,不同廠家的引物在價錢質量上有一些差別,不過價錢一般都是一塊多一個堿基,合成時間約為一周。
這樣的結果是PCR時把整個質粒都給擴出來了,得到的PCR產物是一條鏈完整,另一鏈有缺刻的PCR產物
對于第二個問題:
怎么去掉模板呢?再簡單的方法就是用DpnI酶,DpnI能夠識別甲基化位點并將其酶切,我們用的模板一般都是雙鏈超螺旋質粒,從大腸桿菌里提出來的質粒一般都被甲基化保護起來(除非你用的是甲基化缺陷型的菌株),而PCR產物都是沒有甲基化的,所以DpnI酶能夠特異性地切割模板(質粒)而不會影響PCR產物,從而去掉模板留下PCR產物,所以提質粒時那些菌株一定不能是甲基化缺陷株,不會那么湊巧吧,哈哈。
關于第三個問題:
直接把通過DpnI處理的PCR產物拿去做轉化就行了,呵呵,然后再篩選出陽性克隆,并提出質粒,拿去測序(這個就不用我多說了吧),驗證突變結果,一般都沒問題的啦,我做了幾十個突變了,到目前為止還沒有做不出來的,呵呵,不要砸我啊。
下面講一下具體的實驗步驟以及一些實驗中要注意的事情:
1、 根據現有基因設計引物;
2、 合成引物并準備好模板;
3、 PCR,
4、 DpnI處理酶切產物;
5、 轉化酶切產物;
6、 篩選 陽性克隆;
7、 送測序并測全長。
最后就是慶祝啦,呵呵,沒什么復雜的。
引物和質粒都準備好后,當然就是做PCR嘍,不過對于PCR的酶和buffer,不能用平時的,我們做PCR把整個質粒擴出來,延伸長度達到幾個K,所以要用那些GC buffer或擴增長片段的buffer,另外,要用保真性能較好的PFU酶來擴增,防止引進新的突變。
那種Quick change試劑盒分為幾種不同的類型
什么QuikChange® Site-Directed Mutagenesis Kit標準點突變試劑盒 、QuikChange® XL Site-Directed Mutagenesis Kit長模板單點突變試劑盒(>8kb)從原理上是一樣的,只是PCR的酶和BUFFER不一樣,后面用了比較適合長片段擴增的酶和BUFFER罷了,沒什么特別的東西。另外,DpnI處理的時間最好長一點,最少一個小時吧,最好能有兩三個小時,因為如果模板處理得不干凈,哪怕只有那么一點點,模板直接在平板上長出來,就會導致實驗失敗。
實驗板長出來的菌有兩種可能
一種是質粒DPNI沒處理干凈長出來的(模板),一種是PCR產物轉化出來的
(突變體)
不過這兩種菌長得一模一樣^_^,即使提出質粒來也是一樣(酶切和PCR都無法區分),除了測序,是分不出來的,
做PCR時也最好做一個負對照(不加引物),
實驗管由于PCR時有引物,所以在DNPI處理前里面既含有模板又含有PCR產物,而對照管由于PCR時沒放引物,所以在DPNI處理前里面只有模板。
如果兩者都拿去DNPI處理
就能夠證明模板已經被去除干凈。
若實驗順利的話應該是:正對照長菌負對照不長菌。
如果出現正負對照都長菌,那么就是DpnI沒處理好,
如果正負對照都不長菌,那么有兩種可能,一種是PCR陰性,也就是說PCR出問題了,另外一個可能就是轉化出問題了。要搞清楚是哪個問題,跑膠說明不了問題,那就做個轉化的對照,拿試劑盒的對照實驗去試感受態,馬上就能知道轉化有沒問題。
如果正對照很多菌,負對照有幾個菌,那么就是DPNI處理得不干凈,這個時候就得靠運氣了^_^
大家有什么問題我們可以繼續討論。
另外,如果大家既沒有DpnI酶也沒有好的PCR酶和BUFFER的話,那也有其它辦法進行定點突變,只是麻煩一點,如果大家有需要的話,我會把方法貼上來。
對于多點突變技術及較長片段的缺失插入技術,同樣的,如果大家有需要的話,我會把方法貼上來。
不過,如果你有錢的話,那就去買那個試劑盒吧,其中QuikChange® Site-Directed Mutagenesis Kit標準點突變試劑盒 、QuikChange® XL Site-Directed Mutagenesis Kit長模板單點突變試劑盒(>8kb)的原理我上面已經說了,只是補充了一些我認為的注意事項。如果你更有錢的話,那么你可以叫其它公司幫你做定點突變服務,大約是改一個點1000元左右。如果有需要我可以提供公司的聯系方式。
下面我以一個例子為例來講100個bp以下的堿基插入缺失或者改變實驗方案。其實這種方案并不是那么好的,只不過考慮到大家一般都沒有TYPEII限制性內切酶或者UDG and NTHIII(另外兩種方法),所以才打算先介紹這種方法。
首先先說明一點,這種 方法在原理上存在一定成功機率,也就是說有運氣成分。而定點突變則一般都是百分之百成功的,而這種100bp以下的插入缺失或者堿基改變可能要測幾個克隆才能挑到一個好的克隆,大家如果要用請慎重考慮。
同樣的,我只變那幾十個堿基,并沒有改變載體及其它地方,所以我還是依賴于DPNI酶。
舉例:
Homo sapiens FzE3 是一個人類基因,其含有32個氨基酸的信號肽MRDPGAAAPLSSLGLCALVLALLGALSAGAGA,后面是成熟肽QPYHGEKGISVPDHGFCQPISIPLCTDI
AYNQTILPNLLGHTNQEDAGLEVHQFYPLVKVQCSPELRFFLCSMYAPVCTVLDQAIPPC
RSLCERARQGCEALMNKFGFQWPERLRCENFPVHGAGEICVGQNTSDGSGGPGGGPTAYP
TAPYLPDLPFTALPPGASDGRGRPAFPFSCPRQLKVPPYLGYRFLGERDCGAPCEPGRAN
GLMYFKEEERRFARLWVGVWSVLCCASTLFTVLTYLVDMRRFSYPERPIIFLSGCYFMVA
VAHVAGFLLEDRAVCVERFSDDGYRTVAQGTKKEGCTILFMVLYFFGMASSIWWVILSLT
WFLAAGMKWGHEAIEANSQYFHLAAWAVPAVKTITILAMGQVDGDLLSGVCYVGLSSVDA
LRGFVLAPLFVYLFIGTSFLLAGFVSLFRIRTIMKHDGTKTEKLEKLMVRIGVFSVLYTV
PATIVLACYFYEQAFREHWERTWLLQTCKSYAVPCPPGHFPPMSPDFTVFMIKYLMTMIV
GITTGFWIWSGKTLQSWRRFYHRLSHSSKGETAV,想在信號肽和成熟肽之間插入一個FLAG標簽并在標簽前面加上一個Leucine。即在信號肽和成熟肽之間插入一段序列:TTAATGGACTACAAAGACGATGACGACAAG(一共三十個bp)、
實驗設計:
信號肽:
ATGCGGGACCCCGGCGCGGCCGTTCCGCTTTCGTCCCTGGGCTTCTGTGCCCTGGTGCTG
GCGCTGCTGGGCGCACTGTCCGCGGGCGCCGGGGCG
成熟肽:
CAGCCGTACCACGGAGAGAAGGGC
ATCTCCGTGCCGGACCACGGCTTCTGCCAGCCCATCTCCATCCCGCTGTGCACGGACATC
GCCTACAACCAGACCATCCTGCCCAACCTGCTGGGCCACACGAACCAAGAGGACGCGGGC
CTCGAGGTGCACCAGTTCTACCCGCTGGTGAAGGTGCAGTGTTCTCCCGAACTCCGCTTT
TTCTTATGCTCCATGTATGCGCCCGTGTGCACCGTGCTCGATCAGGCCATCCCGCCGTGT
CGTTCTCTGTGCGAGCGCGCCCGCCAGGGCTGCGAGGCGCTCATGAACAAGTTCGGCTTC
CAGTGGCCCGAGCGCCTGCGCTGCGAGAACTTCCCGGTGCACGGTGCGGGCGAGATCTGC
GTGGGCCAGAACACGTCGGACGGCTCCGGGGGCCCAGGCGGCGGGCCCACTGCCTACCCT
ACCGCGCCCTACCTGCCGGACCTGCCCTTCACCGCGCTGCCCCCGGGGGCCTCAGATGGC
AAGGGGCGTCCCGCCTTCCCCTTCTCATGCCCCCGTCAGCTCAAGGTGCCCCCGTACCTG
GGCTACCGCTTCCTGGGTGAGCGCGATTGTGGCGCCCCGTGCGAACCGGGCCGTGCCAAC
GGCCTGATGTACTTTAAGGAGGAGGAGAGGCGCTTCGCCCGCCTCTGGGTGGGCGTGTGG
TCCGTGCTGTGCTGCGCCTCGACGCTCTTTACCGTTCTCACGTACCTGGTGGACATGCGG
CGCTTCAGCTACCCAGAGCGGCCCATCATCTTCCTGTCGGGCTGCTACTTCATGGTGGCC
GTGGCGCACGTGGCCGGCTTCTTTCTAGAGGACCGCGCCGTGTGCGTGGAGCGCTTCTCG
GACGATGGCTACCGCACGGTGGCGCAGGGCACCAAGAAAGAGGGCTGCACCATCCTCTTC
ATGGTGCTCTACTTCTTCGGCATGGCCAGCTCCATCTGGTGGGTCATTCTGTCTCTCACT
TGGTTCCTGGCGGCCGGCATGAAATGGGGCCACGAAGCCATCGAGGCCAACTCGCAGTAC
TTCCACCTGGCCGCGTGGGCCGTGCCCGCCGTCAAGACCATCACTATCCTGGCCATGGGC
CAGGTAGACGGGGACCTGCTGAACGGGGTGTGCTACGTTGGCTTCTCCAGTGTGGACGCG
CTGCGGGGCTTCGTGCTGGCGCCTCTGTTCGTCTACTTCTTCATAGGCACGTCCTTCTTG
CTGGCCGGCTTCGTGTCCTTCTTCCGTATCCGCACCATCATGAAACACGACGGCACCAAG
ACCGAGAAGCTGGAGAAGCTCATGGTGCGCATCGGCGTCTTCAGCGTGCTCTACACAGTG
CCCGCCACCATCGTCCTGGCCTGCTACTTCTACGAGCAGGCCTTCCGCGAGCACTGGGAG
CGCACCTGGCTCCTGCAGACGTGCAAGAGCTATGCCGTGCCCTGCCCGCCCGGCCACTTC
CCGCCCATGAGCCCCGACTTCACCGTCTTCATGATCAAGTGCCTGATGACCATGATCGTC
GGCATCACCACTGGCTTCTGGATCTGGTCGGGCAAGACCCTGCAGTCGTGGCGCCGCTTC
TACCACAGACTTAGCCACAGCAGCAAGGGAGAGACCGCGGTATGA
插入序列
TTAATGGACTACAAAGACGATGACGACAAG
通過引物3端大于或等于18個堿基的匹配使引物與模板質粒搭配,再通過引物5端的序列來補上那三十個堿基,先用PNK酶把引物磷酸化,再用下面這兩條引物把整個質粒給擴增出來,上游和下游引物就剛好把那三十個堿基給補上了,再參照引物的設計原則做一些潤色,細心的朋友可以具體分析一下這兩條引物。擴出來后再用DPNI酶把模板質粒去掉,再用連接酶把PCR產物的兩端連接起來(雖然是平端連接 ,可是由于是同一條PCR產物的兩端連接,效率會很高),轉化后,測序驗證,OK。
設計引物
forward primer:GGACTACAAAGACGATGACGACAAGCAGCCGTACCACGGAGAGAAG
88.5
reserve primer: ATTAACGCCCCGGCGCCCGCGGACAGT
86.9
但是由于引物的合成是由3端向5端合成,而且每合成多一個堿基的效率最多也是百分之九十九點幾而不是百分之一百,所以我們拿到手的引物其實是一個混合物,比如說我們合成一條長二十個堿基的引物,實際上拿到手的是一個混合物,里面即含有二十個堿基的引物,也含 有一定比率的十九個、十八個、十七個……堿基的引物。
所以我們用這種方法做PCR時,如果連上的是足額長度的引物,那么實驗也就成功了,如果連上的是少一兩個堿基的引物,那么實驗就失敗了,不過引物當中主要的仍是足額長度的引物,所以成功機率還是蠻高的。不過送測序時就要做好準備,可能要測三五個才能拿到一個好的。
如果覺得這樣不好的話,我稍后會附上用TYPEII酶或者UDG,NTHIII做的方法,它們是通過互補粘端來連接,就不存在這個問題。
下面附上詳細實驗過程:
第一步:設計引物;其實只要符合一般引物設計原理就行了,順便說一下,引物一般的話,越長其質量就…………
第二步:引物PNK處理,一般合成的引物其三端是沒有磷酸化的,所以我們要自己進行磷酸化,一般可以讓其磷酸化過夜,不磷酸化的話最后一步連接就連不上哦。
第三步:PCR,跟基因定點突變一樣,要用好的擴增酶和BUFFER,因為要把整個環高保真的壙增出來嘛;
第四步:DPNI處理,跟基因定點突變一樣,要把模板去除干凈。
第五步:連接,加上連接BUFFER和連接酶連接,
第六步:轉化。
定點誘變(DpnI法)
1、引物設計:每條引物都要攜帶有所需的突變位點,引物一般長25~45bp,設計的突變位點需位于引物中部。
2、反應:使用高保真的pyrobest DNA聚合酶 ;循環次數少,一般為12個循環。
反應體系:
10x pyrobest Buffer 5 ul
dNTP Mixture(10mM) 1ul
模板DNA(5~50ng) 1ul
primer 1 (125ng) 1ul
primer 2 (125ng) 1ul
pyrobest DNA polymerase(TaKaRa)(5U/ul) 0.25ul
加無菌蒸餾水至 50ul
3、產物沉淀純化:加1/10 體積的醋酸鈉,1倍體積的異丙醇,混勻置冰上(或-20゜C冰箱)5min,離心棄上清,70~75%乙醇洗鹽兩次,烘干后溶于無菌水中。(此步可省略,直接用 DpnI酶切)
4、DpnI酶切:Buffer 2ul
BSA(100╳) 0.2ul
DNA x ul
DpnI 0.5ul
加無菌去離子水至 20ul
30゜C酶切 1~4 h ;65゜C 水浴15min 終止反應。
5、將酶切產物轉化大腸桿菌DH5a菌株,利用抗生素篩選突變子。
6、測序驗證
把目的基因上面的一個堿基換成另外一個堿基。
二、定點突變的原理
定點突變是指通過聚合酶鏈式反應(PCR)等方法向目的DNA片段(可以是基因組,也可以是質粒)中引入所需變化(通常是表征有利方向的變化),包括堿基的添加、刪除、點突變等。定點突變能迅速、高效的提高DNA所表達的目的蛋白的性狀及表征,是基因研究工作中一種非常有用的手段。
體外定點突變技術是研究蛋白質結構和功能之間的復雜關系的有力工具,也是實驗室中改造/優化基因常用的手段。蛋白質的結構決定其功能,二者之間的關系是蛋白質組研究的重點之一。對某個已知基因的特定堿基進行定點改變、缺失或者插入,可以改變對應的氨基酸序列和蛋白質結構,對突變基因的表達產物進行研究有助于人類了解蛋白質結構和功能的關系,探討蛋白質的結構/結構域。而利用定點突變技術改造基因:比如野生型的綠色熒光蛋白(wtGFP)是在紫外光激發下能夠發出微弱的綠色熒光,經過對其發光結構域的特定氨基酸定點改造,現在的GFP能在可見光的波長范圍被激發(吸收區紅移),而且發光強度比原來強上百倍,甚至還出現了黃色熒光蛋白,藍色熒光蛋白等等。定點突變技術的潛在應用領域很廣,比如研究蛋白質相互作用位點的結構、改造酶的不同活性或者動力學特性,改造啟動子或者DNA作用元件,提高蛋白的抗原性或者是穩定性、活性、研究蛋白的晶體結構,以及藥物研發、基因治療等等方面。
通過設計引物,并利用PCR將模板擴增出來,然后去掉模板,剩下來的就是我們的PCR產物,在PCR產物上就已經把這個點變過來了,然后再轉化,篩選陽性克隆,再測序確定就行了。
三、引物設計原則
引物設計的一般原則不再重復。
突變引物設計的特殊原則:
(1)通常引物長度為25~45 bp,我們建議引物長度為30~35 bp。一般都是以要突變的堿基為中心,加上兩邊的一段序列,兩邊長度至少為11-12 bp。若兩邊引物太短了,很可能會造成突變實驗失敗,因為引物至少要11-12個bp才能與模板搭上,而這種突變PCR要求兩邊都能與引物搭上,所以兩邊最好各設至少12個bp,并且合成多一條反向互補的引物。
(2)如果設定的引物長度為30 bp,接下來需要計算引物的Tm值,看是否達到78℃(GC含量應大于40%)。
(3)如果Tm值低于78℃,則適當改變引物的長度以使其Tm值達到78℃(GC含量應大于40%)。
(4)設計上下游引物時確保突變點在引物的中央位置。
(5)最好使用經過純化的引物。
Tm值計算公式:Tm=0.41×(% of GC)–675/L+81.5
注:L:引物堿基數;% of GC:引物GC含量。
四、引物設計實例
以GCG→ACG為例:
5’-CCTCCTTCAGTATGTAGGCGACTTACTTATTGCGG-3’
(1)首先設計30 bp長的上下游引物,并將A (T)設計在引物的中央位置。
Primer #1: 5’-CCTTCAGTATGTAGACGACTTACTTATTGC-3’
Primer #2: 5’-GCAATAAGTAAGTCGTCTACATACTGAAGG-3’
(2)引物GC含量為40%,L為30,將這兩個數值帶入Tm值計算公式,得到其Tm=75.5(Tm=0.41×40-675/30+81.5)。通過計算可以看出其Tm低于78℃,這樣的引物是不合適的,所以必須調整引物長度。
(3)重新調整引物長度。
Primer #1: 5’-CCTCCTTCAGTATGTAGACGACTTACTTATTGCGG-3’
Primer #2: 5’-CCGCAATAAGTAAGTCGTCTACATACTGAAGGAGG-3’
在引物兩端加5mer(斜體下劃線處),這樣引物的GC含量為45.7%,L值為35,將這兩個數值帶入Tm值計算公式,得到其Tm為80.952(Tm=0.41×47.5-675/35+81.5),這樣的引物就可以用于突變實驗了。
五、突變所用聚合酶及Buffer
引物和質粒都準備好后,當然就是做PCR嘍,不過對于PCR的酶和buffer,不能用平時的,我們做PCR把整個質粒擴出來,延伸長度達到幾個K,所以要用那些GC buffer或擴增長片段的buffer,另外,要用保真性能較好的PFU酶來擴增,防止引進新的突變。
除了使用基因定點突變試劑盒,如Stratagene和塞百盛的試劑盒,但價格昂貴。可以使用高保真的聚合酶,如博大泰克的金牌快速taq酶、Takara的PrimeSTARTM HS DNA polymerase。
六、如何去掉PCR產物
最簡單的方法就是用DpnI酶,DpnI能夠識別甲基化位點并將其酶切,我們用的模板一般都是雙鏈超螺旋質粒,從大腸桿菌里提出來的質粒一般都被甲基化保護起來(除非你用的是甲基化缺陷型的菌株),而PCR產物都是沒有甲基化的,所以DpnI酶能夠特異性地切割模板(質粒)而不會影響PCR產物,從而去掉模板留下PCR產物,所以提質粒時那些菌株一定不能是甲基化缺陷株。
DpnI處理的時間最好長一點,最少一個小時吧,最好能有兩三個小時,因為如果模板處理得不干凈,哪怕只有那么一點點,模板直接在平板上長出來,就會導致實驗失敗。
七、如何拿到質粒
直接把通過DpnI處理的PCR產物拿去做轉化就行了,然后再篩選出陽性克隆,并提出質粒,拿去測序,驗證突變結果。
八、圖示
九、定點突變操作步驟
[A] 誘導突變基因(PCR反應)以待突變的質粒為模板,用設計的引物及Muta-direct™酶進行PCR擴增反應,誘導目的基因突變。
1. 設計點突變引物。
[注]參考引物設計指導
2. 準備模板質粒DN A
[注]用dam+型菌株(例如DH5α菌株)作為宿主菌。在end+型菌株中常有克隆數低的現象,但是對突變效率沒有影響。提取質粒DNA時我們建議您使用本公司的質粒提純試劑盒。
3. [選項]對照反應體系(50μl反應體系)
| 10×Reaction Buffer | 5μl |
| pUC18 control plasmid(10ng/μl,total 20ng) | 2μl |
| Control primer mix(20pmol/μl) | 2μl |
| dNTP mixture(each 2.5mM) | 2μl |
| dH2O | 38μl |
| Muta-direct™ Enzyme | 1μl |
| 10×Reaction Buffer | 5μl |
| Sample plasmid(10ng/μl,total 20ng) | 2μl |
| Sample primer (F)(10pmol/μl) | 1μl |
| Sample primer (R)(10pmol/μl) | 1μl |
| dNTP mixture(each 2.5mM) | 2μl |
| dH2O | 38μl |
| Muta-direct™ Enzyme | 1μl |
[注]按如下參數設置PCR擴增條件。
| Cycles | Temperature | Reaction Time |
| 1cycle | 95℃ | 30sec |
| 15cycle | 95℃ | 30sec |
| 55℃ | 1min | |
| 72℃ | 1min per plasmid Kb |
[注] 按下列提供的PCR條件進行擴增,控制PCR循環數。注意當突變點位點超過4個時會發生突變率降低的現象。
| Mutation | Cycles |
| 1~2Nucleotide | 15cycles |
| 3Nucleotides | 18cycles |
PCR反應結束后使用Mutazyme™酶消化甲基化質粒從而選擇突變質粒DNA。
1. 準備PCR反應產物
2. 加入1μl(10U/μl)Mutazyme™酶37℃溫育1小時。
[注]當質粒DNA用量過多時Mutazyme™酶可能發生與樣品反應不完全的現象。因此我們建議為了保證突變率請嚴格遵照實驗步驟進行操作。如果突變率低,可以適當延長反應時間或增加Mutazyme™酶用量。
[C]轉化
反應完畢后在質粒DNA上會產生缺口,當把這個質粒DNA轉入E.coli中時請選擇dam+型菌株,例如DH5α。
1. 將10μl樣品加到50μl感受態細胞里,然后放置在冰上30分鐘。
2. 接下來可以參照一般的轉化步驟進行。
序列分析
通常當LB平板上出白色菌落則表明發生了突變。
為了證實這一結果,我們建議對白色單菌落進行測序分析。
先講最簡單的一個點的定點突變技術,其它較長片段的突變,刪除,插入技術以后會慢慢奉上:在做實驗之前,我們首先要搞清楚實驗的目的和實驗的原理。
實驗的目的應該比較明確吧:就是要把自己的基因上面的一個堿基換成另外一個堿基。一般情況下我們會有幾種可能使我們需要這樣去做:
第一:我們吊出來的基因有點突變,相信這可能是大家經常會遇到的問題。基因好不容易吊出來,并裝進了自己的載體,卻發現有一兩個堿基跟自己的預期序列或所有的公共數據庫不匹配,然后暴昏。
大家實驗室里面還是用Taq酶為主吧,Pfu這樣的高保真酶大家應該用得不多吧,Taq酶的優點和缺點都很明顯:優點就是擴增效能強,缺點就是保真性差,其錯配機率是比較高的,相關數字忘了,大家可以去網上查那個數字,不過感覺如果是2000bp的基因,如果擴四五十個循環的話,很大機率會出現點突變,當然這也跟具體PCR體系里的Buffer有很大關系,詳細情況這里就不討論了。
第二:要研究基因的功能,在基因上自己選定位置更換堿基的保守序列,或者改造成不同的亞型,總之就是要人工改造堿基序列符合自己的實驗需要,相信這也是那些研究基因的人經常的一種思路吧。
對于第一種情況:我們首先要分析出現堿基不匹配的位置是不是重要的位置,如果不是很重要,大可不必管它,比如說是三聯密碼子的最后一位,堿基的改變并沒有引起相應氨基酸的改變,那么一般情況下也可以不去理它。另外,在NCBI上人類的基因的版本一直在變化,也就是說同一個基因有不同的版本,或者稱不同的亞型,其堿基序列有些許的差異,只要自己克隆出來的堿基序列與其中一個相匹配,一般也就可以不做定點突變了。如果有時間沒錢,那干脆重新PCR然后再克隆進自己的載體了,不過最好換個保真性好一點的酶如PFU,或者PCR循環數低一點,不過這些東西有時候也得靠運氣啦。實在不行的話再來做定點突變。
對于第二種情況:這種情況下一般也就只能做定點突變了。
接下來開始聊一聊定點突變的原理吧,那個Stratagene試劑盒!上面有一個說明書,說得好像很正規,不過上面好多都是什么專利啊什么注意之類的話,看都不看,我們簡明扼要地只講實驗方面,通過設計引物,并利用PCR將模板擴增出來,然后去掉模板,剩下來的就是我們的PCR產物,在PCR產物上就已經把這個點變過來了,然后再轉化,篩選陽性克隆,再測序確定就行了。
大家馬上就會想到幾個問題了:
第一:引物怎么設計呢?
第二:模板怎么去掉呢?
第三:怎么拿到質粒呢?
對于第一個問題:怎么設計引物?
我只能講一些原則,并舉一些例子。
引物設計的原則其它貼子上都有講,這里就不重復了:
不過這種突變引物要加上一個原則:
一般都是以要突變的堿基為中心,加上兩邊的一段序列,兩邊長度至少為11-12base pair。
若兩邊引物太短了,很可能會造成突變實驗失敗,大家應該都知道,引物至少要11-12個base pair才能與模板搭上,而這種突變PCR要求兩邊都能與引物搭上,所以兩邊最好各設至少12個base pair,并且合成多一條反向互補的引物。
這么說大家可能不是很清楚,那我就舉個例子吧:
X71661.1 TATCAGGAGGAATTTGAGCACTTTCAACAAGAATTGGATAAAAAAAAAGAGGAATTCCAG 960
現有序列 TATCAGGAGGAATTTGAGCACTTTCAACAAGAATTGGATAAAAAAAAAGAGGAATTCCAG 924
*********************************************** ************
|----deletion
X71661.1 AAGGGCCACCCCGACCTCCAAGGGCAGCCTGCGGAGGAAATATTTGAGAGTGTAGGAGAT 1020
現有序列 AAGGGCCACCCCGACCTCCAAGGGCAGCCTGCGGAGGAAATATTTGAGAGTGTAGGAGAT 984(上面為目的序列,下面為現有序列:我們發現有一個A堿基的缺失,其直接結果是在表達蛋白時后面的氨基酸全部錯配)
我們以它為中心設計引物:兩邊各至少12個堿基,左邊由于含有較多的A造成引物GC%含量過低,故拉長引物使GC%含量不至過低,也使引物退火溫度升高。
故合成引物CAACAAGAATTGGATAAAAAAAAAGAGGAATTCCAGAAG
并合成反向互補引物CTTCTGGAATTCCTCTTTTTTTTTATCCAATTCTTGTTG
其實也不一定要反向互補序列,只要反向引物也是兩邊都有大于12個堿基,同時符合引物設計的原則就行了。
引物合成公司有很多家,大家可以去尋找,不同廠家的引物在價錢質量上有一些差別,不過價錢一般都是一塊多一個堿基,合成時間約為一周。
這樣的結果是PCR時把整個質粒都給擴出來了,得到的PCR產物是一條鏈完整,另一鏈有缺刻的PCR產物
對于第二個問題:
怎么去掉模板呢?再簡單的方法就是用DpnI酶,DpnI能夠識別甲基化位點并將其酶切,我們用的模板一般都是雙鏈超螺旋質粒,從大腸桿菌里提出來的質粒一般都被甲基化保護起來(除非你用的是甲基化缺陷型的菌株),而PCR產物都是沒有甲基化的,所以DpnI酶能夠特異性地切割模板(質粒)而不會影響PCR產物,從而去掉模板留下PCR產物,所以提質粒時那些菌株一定不能是甲基化缺陷株,不會那么湊巧吧,哈哈。
關于第三個問題:
直接把通過DpnI處理的PCR產物拿去做轉化就行了,呵呵,然后再篩選出陽性克隆,并提出質粒,拿去測序(這個就不用我多說了吧),驗證突變結果,一般都沒問題的啦,我做了幾十個突變了,到目前為止還沒有做不出來的,呵呵,不要砸我啊。
下面講一下具體的實驗步驟以及一些實驗中要注意的事情:
1、 根據現有基因設計引物;
2、 合成引物并準備好模板;
3、 PCR,
4、 DpnI處理酶切產物;
5、 轉化酶切產物;
6、 篩選 陽性克隆;
7、 送測序并測全長。
最后就是慶祝啦,呵呵,沒什么復雜的。
引物和質粒都準備好后,當然就是做PCR嘍,不過對于PCR的酶和buffer,不能用平時的,我們做PCR把整個質粒擴出來,延伸長度達到幾個K,所以要用那些GC buffer或擴增長片段的buffer,另外,要用保真性能較好的PFU酶來擴增,防止引進新的突變。
那種Quick change試劑盒分為幾種不同的類型
什么QuikChange® Site-Directed Mutagenesis Kit標準點突變試劑盒 、QuikChange® XL Site-Directed Mutagenesis Kit長模板單點突變試劑盒(>8kb)從原理上是一樣的,只是PCR的酶和BUFFER不一樣,后面用了比較適合長片段擴增的酶和BUFFER罷了,沒什么特別的東西。另外,DpnI處理的時間最好長一點,最少一個小時吧,最好能有兩三個小時,因為如果模板處理得不干凈,哪怕只有那么一點點,模板直接在平板上長出來,就會導致實驗失敗。
實驗板長出來的菌有兩種可能
一種是質粒DPNI沒處理干凈長出來的(模板),一種是PCR產物轉化出來的
(突變體)
不過這兩種菌長得一模一樣^_^,即使提出質粒來也是一樣(酶切和PCR都無法區分),除了測序,是分不出來的,
做PCR時也最好做一個負對照(不加引物),
實驗管由于PCR時有引物,所以在DNPI處理前里面既含有模板又含有PCR產物,而對照管由于PCR時沒放引物,所以在DPNI處理前里面只有模板。
如果兩者都拿去DNPI處理
就能夠證明模板已經被去除干凈。
若實驗順利的話應該是:正對照長菌負對照不長菌。
如果出現正負對照都長菌,那么就是DpnI沒處理好,
如果正負對照都不長菌,那么有兩種可能,一種是PCR陰性,也就是說PCR出問題了,另外一個可能就是轉化出問題了。要搞清楚是哪個問題,跑膠說明不了問題,那就做個轉化的對照,拿試劑盒的對照實驗去試感受態,馬上就能知道轉化有沒問題。
如果正對照很多菌,負對照有幾個菌,那么就是DPNI處理得不干凈,這個時候就得靠運氣了^_^
大家有什么問題我們可以繼續討論。
另外,如果大家既沒有DpnI酶也沒有好的PCR酶和BUFFER的話,那也有其它辦法進行定點突變,只是麻煩一點,如果大家有需要的話,我會把方法貼上來。
對于多點突變技術及較長片段的缺失插入技術,同樣的,如果大家有需要的話,我會把方法貼上來。
不過,如果你有錢的話,那就去買那個試劑盒吧,其中QuikChange® Site-Directed Mutagenesis Kit標準點突變試劑盒 、QuikChange® XL Site-Directed Mutagenesis Kit長模板單點突變試劑盒(>8kb)的原理我上面已經說了,只是補充了一些我認為的注意事項。如果你更有錢的話,那么你可以叫其它公司幫你做定點突變服務,大約是改一個點1000元左右。如果有需要我可以提供公司的聯系方式。
下面我以一個例子為例來講100個bp以下的堿基插入缺失或者改變實驗方案。其實這種方案并不是那么好的,只不過考慮到大家一般都沒有TYPEII限制性內切酶或者UDG and NTHIII(另外兩種方法),所以才打算先介紹這種方法。
首先先說明一點,這種 方法在原理上存在一定成功機率,也就是說有運氣成分。而定點突變則一般都是百分之百成功的,而這種100bp以下的插入缺失或者堿基改變可能要測幾個克隆才能挑到一個好的克隆,大家如果要用請慎重考慮。
同樣的,我只變那幾十個堿基,并沒有改變載體及其它地方,所以我還是依賴于DPNI酶。
舉例:
Homo sapiens FzE3 是一個人類基因,其含有32個氨基酸的信號肽MRDPGAAAPLSSLGLCALVLALLGALSAGAGA,后面是成熟肽QPYHGEKGISVPDHGFCQPISIPLCTDI
AYNQTILPNLLGHTNQEDAGLEVHQFYPLVKVQCSPELRFFLCSMYAPVCTVLDQAIPPC
RSLCERARQGCEALMNKFGFQWPERLRCENFPVHGAGEICVGQNTSDGSGGPGGGPTAYP
TAPYLPDLPFTALPPGASDGRGRPAFPFSCPRQLKVPPYLGYRFLGERDCGAPCEPGRAN
GLMYFKEEERRFARLWVGVWSVLCCASTLFTVLTYLVDMRRFSYPERPIIFLSGCYFMVA
VAHVAGFLLEDRAVCVERFSDDGYRTVAQGTKKEGCTILFMVLYFFGMASSIWWVILSLT
WFLAAGMKWGHEAIEANSQYFHLAAWAVPAVKTITILAMGQVDGDLLSGVCYVGLSSVDA
LRGFVLAPLFVYLFIGTSFLLAGFVSLFRIRTIMKHDGTKTEKLEKLMVRIGVFSVLYTV
PATIVLACYFYEQAFREHWERTWLLQTCKSYAVPCPPGHFPPMSPDFTVFMIKYLMTMIV
GITTGFWIWSGKTLQSWRRFYHRLSHSSKGETAV,想在信號肽和成熟肽之間插入一個FLAG標簽并在標簽前面加上一個Leucine。即在信號肽和成熟肽之間插入一段序列:TTAATGGACTACAAAGACGATGACGACAAG(一共三十個bp)、
實驗設計:
信號肽:
ATGCGGGACCCCGGCGCGGCCGTTCCGCTTTCGTCCCTGGGCTTCTGTGCCCTGGTGCTG
GCGCTGCTGGGCGCACTGTCCGCGGGCGCCGGGGCG
成熟肽:
CAGCCGTACCACGGAGAGAAGGGC
ATCTCCGTGCCGGACCACGGCTTCTGCCAGCCCATCTCCATCCCGCTGTGCACGGACATC
GCCTACAACCAGACCATCCTGCCCAACCTGCTGGGCCACACGAACCAAGAGGACGCGGGC
CTCGAGGTGCACCAGTTCTACCCGCTGGTGAAGGTGCAGTGTTCTCCCGAACTCCGCTTT
TTCTTATGCTCCATGTATGCGCCCGTGTGCACCGTGCTCGATCAGGCCATCCCGCCGTGT
CGTTCTCTGTGCGAGCGCGCCCGCCAGGGCTGCGAGGCGCTCATGAACAAGTTCGGCTTC
CAGTGGCCCGAGCGCCTGCGCTGCGAGAACTTCCCGGTGCACGGTGCGGGCGAGATCTGC
GTGGGCCAGAACACGTCGGACGGCTCCGGGGGCCCAGGCGGCGGGCCCACTGCCTACCCT
ACCGCGCCCTACCTGCCGGACCTGCCCTTCACCGCGCTGCCCCCGGGGGCCTCAGATGGC
AAGGGGCGTCCCGCCTTCCCCTTCTCATGCCCCCGTCAGCTCAAGGTGCCCCCGTACCTG
GGCTACCGCTTCCTGGGTGAGCGCGATTGTGGCGCCCCGTGCGAACCGGGCCGTGCCAAC
GGCCTGATGTACTTTAAGGAGGAGGAGAGGCGCTTCGCCCGCCTCTGGGTGGGCGTGTGG
TCCGTGCTGTGCTGCGCCTCGACGCTCTTTACCGTTCTCACGTACCTGGTGGACATGCGG
CGCTTCAGCTACCCAGAGCGGCCCATCATCTTCCTGTCGGGCTGCTACTTCATGGTGGCC
GTGGCGCACGTGGCCGGCTTCTTTCTAGAGGACCGCGCCGTGTGCGTGGAGCGCTTCTCG
GACGATGGCTACCGCACGGTGGCGCAGGGCACCAAGAAAGAGGGCTGCACCATCCTCTTC
ATGGTGCTCTACTTCTTCGGCATGGCCAGCTCCATCTGGTGGGTCATTCTGTCTCTCACT
TGGTTCCTGGCGGCCGGCATGAAATGGGGCCACGAAGCCATCGAGGCCAACTCGCAGTAC
TTCCACCTGGCCGCGTGGGCCGTGCCCGCCGTCAAGACCATCACTATCCTGGCCATGGGC
CAGGTAGACGGGGACCTGCTGAACGGGGTGTGCTACGTTGGCTTCTCCAGTGTGGACGCG
CTGCGGGGCTTCGTGCTGGCGCCTCTGTTCGTCTACTTCTTCATAGGCACGTCCTTCTTG
CTGGCCGGCTTCGTGTCCTTCTTCCGTATCCGCACCATCATGAAACACGACGGCACCAAG
ACCGAGAAGCTGGAGAAGCTCATGGTGCGCATCGGCGTCTTCAGCGTGCTCTACACAGTG
CCCGCCACCATCGTCCTGGCCTGCTACTTCTACGAGCAGGCCTTCCGCGAGCACTGGGAG
CGCACCTGGCTCCTGCAGACGTGCAAGAGCTATGCCGTGCCCTGCCCGCCCGGCCACTTC
CCGCCCATGAGCCCCGACTTCACCGTCTTCATGATCAAGTGCCTGATGACCATGATCGTC
GGCATCACCACTGGCTTCTGGATCTGGTCGGGCAAGACCCTGCAGTCGTGGCGCCGCTTC
TACCACAGACTTAGCCACAGCAGCAAGGGAGAGACCGCGGTATGA
插入序列
TTAATGGACTACAAAGACGATGACGACAAG
通過引物3端大于或等于18個堿基的匹配使引物與模板質粒搭配,再通過引物5端的序列來補上那三十個堿基,先用PNK酶把引物磷酸化,再用下面這兩條引物把整個質粒給擴增出來,上游和下游引物就剛好把那三十個堿基給補上了,再參照引物的設計原則做一些潤色,細心的朋友可以具體分析一下這兩條引物。擴出來后再用DPNI酶把模板質粒去掉,再用連接酶把PCR產物的兩端連接起來(雖然是平端連接 ,可是由于是同一條PCR產物的兩端連接,效率會很高),轉化后,測序驗證,OK。
設計引物
forward primer:GGACTACAAAGACGATGACGACAAGCAGCCGTACCACGGAGAGAAG
88.5
reserve primer: ATTAACGCCCCGGCGCCCGCGGACAGT
86.9
但是由于引物的合成是由3端向5端合成,而且每合成多一個堿基的效率最多也是百分之九十九點幾而不是百分之一百,所以我們拿到手的引物其實是一個混合物,比如說我們合成一條長二十個堿基的引物,實際上拿到手的是一個混合物,里面即含有二十個堿基的引物,也含 有一定比率的十九個、十八個、十七個……堿基的引物。
所以我們用這種方法做PCR時,如果連上的是足額長度的引物,那么實驗也就成功了,如果連上的是少一兩個堿基的引物,那么實驗就失敗了,不過引物當中主要的仍是足額長度的引物,所以成功機率還是蠻高的。不過送測序時就要做好準備,可能要測三五個才能拿到一個好的。
如果覺得這樣不好的話,我稍后會附上用TYPEII酶或者UDG,NTHIII做的方法,它們是通過互補粘端來連接,就不存在這個問題。
下面附上詳細實驗過程:
第一步:設計引物;其實只要符合一般引物設計原理就行了,順便說一下,引物一般的話,越長其質量就…………
第二步:引物PNK處理,一般合成的引物其三端是沒有磷酸化的,所以我們要自己進行磷酸化,一般可以讓其磷酸化過夜,不磷酸化的話最后一步連接就連不上哦。
第三步:PCR,跟基因定點突變一樣,要用好的擴增酶和BUFFER,因為要把整個環高保真的壙增出來嘛;
第四步:DPNI處理,跟基因定點突變一樣,要把模板去除干凈。
第五步:連接,加上連接BUFFER和連接酶連接,
第六步:轉化。
定點誘變(DpnI法)
1、引物設計:每條引物都要攜帶有所需的突變位點,引物一般長25~45bp,設計的突變位點需位于引物中部。
2、反應:使用高保真的pyrobest DNA聚合酶 ;循環次數少,一般為12個循環。
反應體系:
10x pyrobest Buffer 5 ul
dNTP Mixture(10mM) 1ul
模板DNA(5~50ng) 1ul
primer 1 (125ng) 1ul
primer 2 (125ng) 1ul
pyrobest DNA polymerase(TaKaRa)(5U/ul) 0.25ul
加無菌蒸餾水至 50ul
3、產物沉淀純化:加1/10 體積的醋酸鈉,1倍體積的異丙醇,混勻置冰上(或-20゜C冰箱)5min,離心棄上清,70~75%乙醇洗鹽兩次,烘干后溶于無菌水中。(此步可省略,直接用 DpnI酶切)
4、DpnI酶切:Buffer 2ul
BSA(100╳) 0.2ul
DNA x ul
DpnI 0.5ul
加無菌去離子水至 20ul
30゜C酶切 1~4 h ;65゜C 水浴15min 終止反應。
5、將酶切產物轉化大腸桿菌DH5a菌株,利用抗生素篩選突變子。
6、測序驗證
相關技術服務
推薦閱讀
最新動態
-
09.23
中藥的現代詮釋:外泌體如何革新傳統醫學?
-
07.02
1+1>2!深度解析RNA測序數據挖掘邏輯和后期實驗設計思路,輕松研獲10+ SCI
-
07.01
“稻”亦有道——盤點近期水稻研究的重大突破
-
06.28
科學與美學的結合體:植物亞細胞定位技術詳解
-
06.28
“聚焦新質生產力,激發科研新動能”|LCA躋身蛋白互作研究的新銳力量
-
06.05
知無不“研”|一文讀懂免疫共沉淀技術(Co-IP)
-
05.14
四大研究利器(Co-IP、BIFC、Y2H、GST pull-down)助力速配蛋白互作“最佳拍檔”
-
05.14
高效、精準、直觀、實時——取經“蛋白互作研究翹楚”BIFC!
-
05.14
轉染效率低、干擾效果差、重復性欠佳...siRNA研究頻遇“攔路虎”怎么辦?
-
04.22
一文讀懂EMSA技術核心要點,讓“emsa” 秒變“easy”
 X
X 






