
多組學聯合分析研究腸道微生物群促進小鼠肝臟脂肪酸去飽和和延長
信息來源:金開瑞 作者:genecreate 發布時間:2018-10-12 13:45:11
題目:The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice
多組學聯合分析研究腸道微生物群促進小鼠肝臟脂肪酸去飽和和延長
期刊:Nature Communications
影響因子:12.353
主要技術:轉錄組、蛋白質組、磷酸化蛋白質組、脂質組、16sRNA測序
研究背景
哺乳動物腸道具有高度復雜的腸道微生物群落,在過去的幾十年中,許多小鼠模型已被用于研究腸道微生物群及其與宿主的關系。本研究整合轉錄組學、蛋白質組學、磷酸化蛋白質組學和脂質組學進行分析,提出了腸道微生物對肝臟脂質代謝的影響。
研究結果
1. 轉錄組和蛋白質組聯合分析
作者針對無菌小鼠模型(GF)和無特定病原體小鼠模型(SPF)的肝臟和血漿樣品進行多組學分析。其中,小鼠肝臟樣本的轉錄組使用微陣列芯片技術分析,總共分析了41174個探針,其中32308個可以定位到轉錄本,7769個探針顯著差異表達(3937個基因),GF和SPF之間的差異倍數最大達到14.3(圖1a)。通路富集分析揭示了GF小鼠中顯著富集的多個代謝通路,如藥物代謝、脂質代謝(圖1b)。
使用TMT技術對小鼠肝臟樣本進行蛋白質組學檢測分析,共鑒定到5875個蛋白質,GF和SPF中存在顯著差異的蛋白數目為455(圖1c),差異倍數最高達3.7。差異表達最顯著的蛋白質分別為L3HYPDH、CYP3A11、EPHX1、ACSF2、SCD1,這幾個重要的蛋白質均參與了特定的代謝過程,如亞油酸和視黃醇代謝(圖1d)。
作者使用基因名稱合并了蛋白質組和轉錄組數據,以便識別在兩個組學層中上調或下調的基因。兩次組學同時鑒定到4843個基因(1i),其中2822個基因(約58%)為GF和SPF共有,984個基因在SPF中持續高表達(圖1e,Q2),1,838個基因在GF中持續高表達(圖1e,Q4)。蛋白質組和轉錄組基因功能富集分析顯示SPF小鼠中上調的基因(Q2)在脂質代謝中參與的8個GO生物過程中有5個表現出富集(圖1f),下調基因(Q4)則沒有顯著的GO注釋。作者發現,影響脂質代謝的關鍵酶,如ACLY、FASN、SCD1和ELOVL6,在轉錄組與蛋白質組中均存在差異表達。
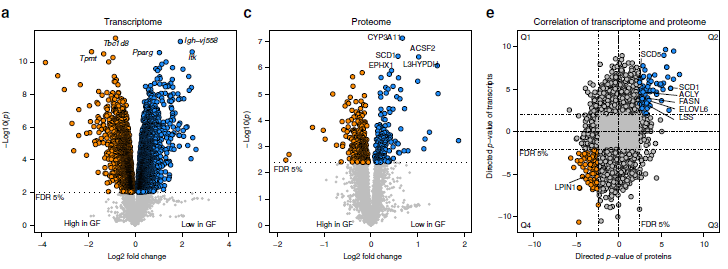
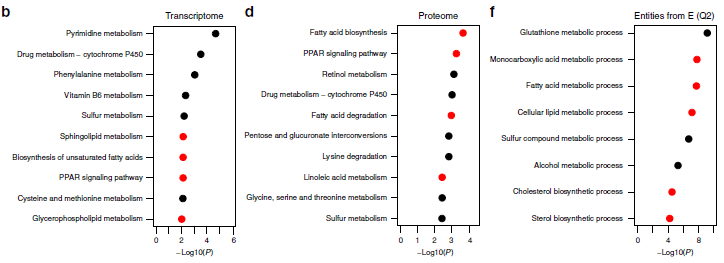
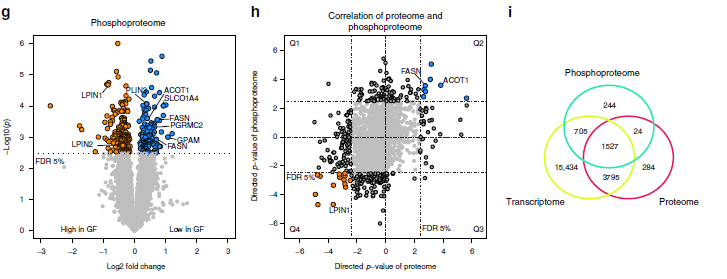
圖1 SPF和GF小鼠肝臟樣品的轉錄組學和蛋白質組學分析
2. 磷酸化蛋白質組分析
蛋白質磷酸化在酶的活化和失活中起重要作用,因此,作者同時對GF和SPF小鼠的肝臟樣品進行磷酸化蛋白質組分析(磷酸化TMT)。在本次試驗中,共鑒定到了5558種磷酸化肽段,其中1551個磷酸肽段,還可以檢測相應的蛋白質,轉錄組、蛋白質組和磷酸化蛋白質組共同檢測到1527個磷酸肽段(圖1i)。作者研究發現363種蛋白質在GF和SPF動物之間具有顯著不同的磷酸化水平(圖1g),其中,8種蛋白參與到了脂質代謝ACOT1、FASN、GPAM、LPIN1 / 2、PGRMC2、PLIN3和SLCO1A4。
3. 脂質組學分析
為了檢測脂肪酸和脂質代謝中涉及的酶的變化是否導致GF小鼠中的脂質水平改變,作者進行了全面的定量脂質組分析。使用氣相色譜-質譜聯用(GC-MS)技術共檢測到血漿與肝臟樣本中的20類525種脂質,如脂肪酸(FAs)、甘油磷脂、鞘脂和甾醇等。作者研究發現肝臟樣本中的16種脂質在GF和SPF小鼠之間差異顯著,血漿樣本中則15種脂質差異顯著。最重要的是,作者發現SPF和GF小鼠之間存在從單不飽和脂質向多不飽和脂質系統轉變的趨勢。SPF小鼠樣品中的棕櫚油酸和含有單不飽和酰基鏈的甘油磷脂,含量高出1.5倍(圖2a-d),而在GF小鼠中,多不飽和脂肪酸和甘油磷脂(如花生四烯酸)的含量較高。磷脂酰膽堿(PC,37%)、磷脂酰乙醇胺(PE,19%)、游離膽固醇(FC,11%)是肝臟中的主要脂質類別(圖2e),鞘脂僅代表肝臟脂質的一小部分,約占2%(圖2f)。血漿脂質則以磷脂酰膽堿(PC,27%)、膽固醇酯(CE,54%)和游離膽固醇(FC,10%)為主(圖2g),而且肝臟和血漿樣品中每種脂質類別的總濃度沒有差異(圖2e,g)。
總而言之,作者認為以上數據顯示腸道微生物群影響肝臟和血漿中的特定脂肪酸和甘油磷脂物種譜。含有腸道微生物群的小鼠會較高比例的含有單不飽和脂肪酸(MUFA),而GF小鼠以脂質飽和脂肪酸(SAFA) 和多不飽和脂肪酸(PUFA)為主。
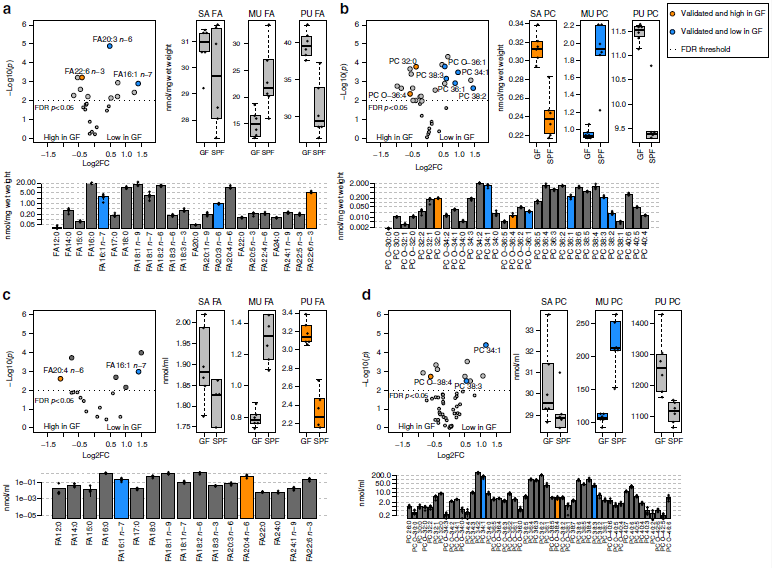
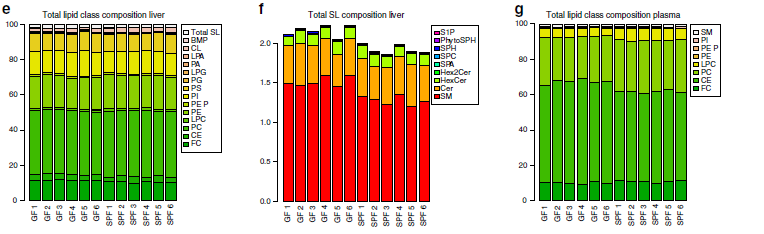
圖2 GF和SPF小鼠肝臟和血漿的定量脂質組分析
4. 組學數據重建脂肪酸代謝
作者假設GF小鼠的脂肪酸飽和度和延伸能力與SPF小鼠不同,導致肝臟甘油磷脂酰基鏈譜改變。因此,作者重建了脂肪酸代謝途徑并整合了磷酸化蛋白質組、蛋白質組和轉錄組,系統地概述肝臟脂質代謝過程(圖3a,b)。作者在3個特定的脂肪酸轉化步驟中觀察到GF和SPF小鼠之間的顯著差異:(I)借助ACAC A / B和FASN,從乙酰-CoA從頭合成脂肪酸的過程(圖3a); (II)通過SCD1將棕櫚酸酯(FA16:0)去飽和為棕櫚油酸酯(FA16:1 n-7)(圖3a); (III)通過ELOVL5(圖4b)將FA18:3 n-6延伸至FA20:3 n-6(圖3b)。在所有步驟中,SPF具有較高值。為了支持(II)和(III),作者將脂肪酸產物/前體比率與相應的mRNA或蛋白質豐度相關聯。如圖 3d所示 ,SCD1表達與16:1 n -7 / FA16:0比率(mRNA: R = 0.93; p <0.00002;蛋白質:R = 0.94; p <0.00005)強相關,ELOVL5表達與20:3 n -6/18:3 n -6的比率(mRNA: R = 0.79; p <0.002; 蛋白質:未檢測到)強相關。此外,作者還發現脂肪酸部分的反應比(產物/前體水平)與PC和LPC物種的相應去飽和度和伸長指數高度相關(圖3e),表明改變的FA代謝過程清楚地反映在酰基中。
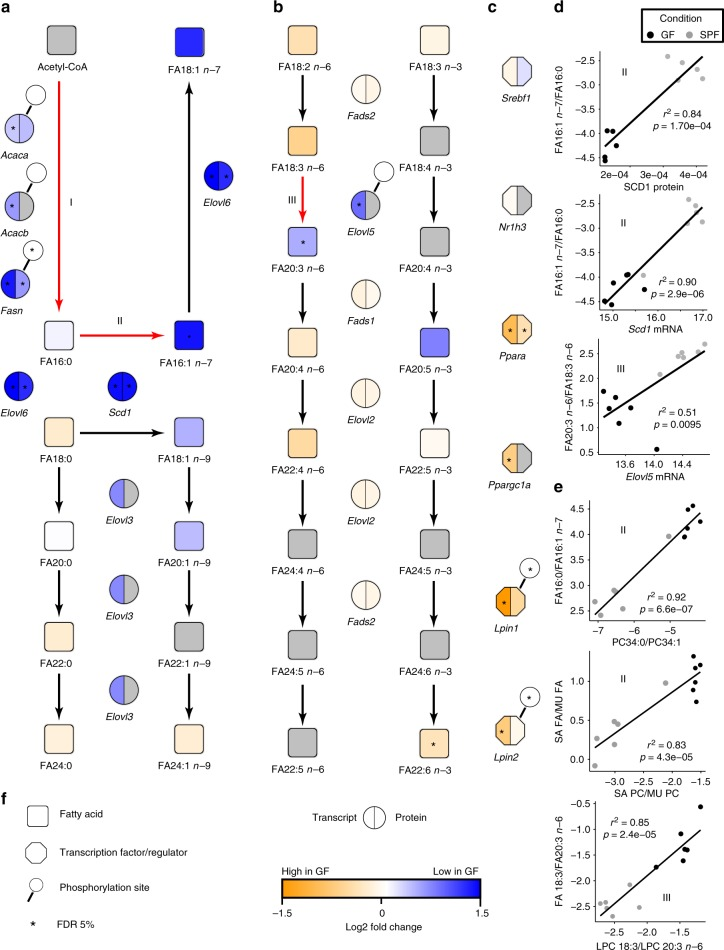
圖3 多組學數據重建脂肪酸代謝途徑
5. 基于脂肪酸譜的綜合分類
為了獲得腸道微生物群特異性調節脂質代謝過程的進一步證據,作者利用短期抗生素(氨芐青霉素(A)、萬古霉素(V)、甲硝唑(M)、或萬古霉素(V)和甲硝唑(M)的組合)處理SPF的腸道微生物生態系統,GC-MS定量肝臟脂肪酸譜,作者發現通過FA 16:0、FA 20:3 n-6、FA 20:4 n-6和FA 22:6 n-3進行分類,既能清楚地將GF與SPF小鼠區分開(圖4a,b),也可將不同抗生素處理與未處理的小鼠區分開(圖4c)。Scd1和Elovl5的mRNA表達水平明顯遵循分類評分的趨勢,低評分樣本有低表達水平(圖4d)。這些結果支持了腸道微生物群影響脂肪酸代謝代謝,特別是由SCD1和ELOVL5產生的脂肪酸(圖3a,b;反應II和III)。
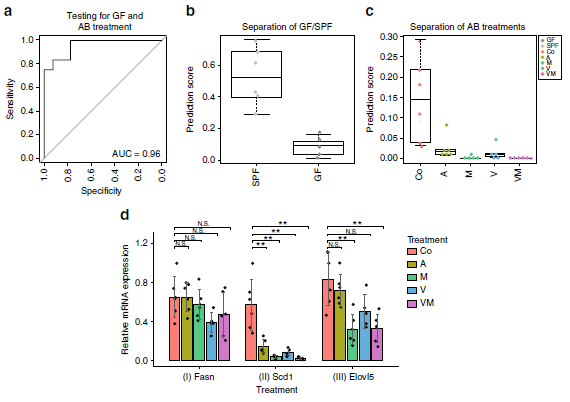
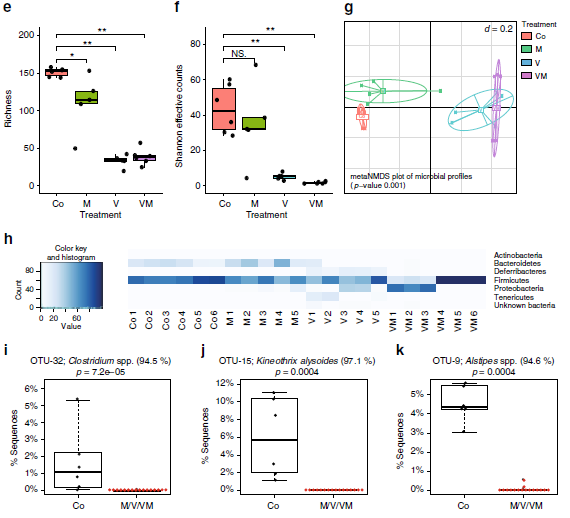
圖4抗生素對腸道微生物種類的影響
6. 腸道微生物群組成分析
為了確定抗生素對腸道微生物群多樣性和組成的影響,作者通過高通量測序分析了16S rRNA基因的V3-V4擴增子。本次實驗共檢測總到376071個序列,代表172個OTU(每個樣品99±49 OTU)。所有抗生素均影響α多樣性(圖4e,f),與單獨的M相比,V和VM的組合具有特別顯著的影響,β多樣性分析揭示了V和VM樣品的顯著聚集,這些樣品與對照組分開,但是彼此并不分離(圖4g),受抗生素影響最大的OUT來自于厚壁菌和擬桿菌。
文章小結
宿主的腸道微生物與脂質穩態之間的相互作用,與宿主的生理學機能和代謝疾病高度相關。本研究整合轉錄組學、蛋白質組學、磷酸化蛋白質組學和脂質組學進行分析,提出了腸道微生物促進肝臟脂質代謝的多組學觀點。微生物可以通過硬脂酰輔酶A脫氫酶1(SCD1)誘導單不飽和脂肪酸生成,并通過脂肪酸延長酶5誘導多不飽和脂肪酸延伸,導致甘油磷脂酰基鏈分布的顯著改變。
解析文獻
Alida Kindt, Gerhard Liebisch, et al. The gut microbiota promotes hepatic fatty acid desaturation and elongation in mice. Nature Communications, 2018, DOI: 10.1038/s41467-018-05767-4
參考文獻
1. Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics usingStageTips. Nat. Protoc, 2007, 2, 1896–1906.
2. Ruprecht, B. et al. Evaluation of kinase activity profiling using chemical proteomics. ACS Chem. Biol. 2015, 10, 2743–2752.
3. Bartram, J. et al. Accurate sample assignment in a multiplexed, ultrasensitive, high-throughput sequencing assay for minimal residual disease. J. Mol. Diagn.2016, 18, 494–506.
4. Ruprecht, B., Zecha, J., Zolg, D. P. & Kuster, B. High pH reversed-phase microcolumns for simple, sensitive, and efficient fractionation of proteome and (TMT labeled) phosphoproteome digests. Methods Mol. Biol. 2017, 1550, 83–98.
最新動態
-
09.23
中藥的現代詮釋:外泌體如何革新傳統醫學?
-
07.02
1+1>2!深度解析RNA測序數據挖掘邏輯和后期實驗設計思路,輕松研獲10+ SCI
-
07.01
“稻”亦有道——盤點近期水稻研究的重大突破
-
06.28
科學與美學的結合體:植物亞細胞定位技術詳解
-
06.28
“聚焦新質生產力,激發科研新動能”|LCA躋身蛋白互作研究的新銳力量
-
06.05
知無不“研”|一文讀懂免疫共沉淀技術(Co-IP)
-
05.14
四大研究利器(Co-IP、BIFC、Y2H、GST pull-down)助力速配蛋白互作“最佳拍檔”
-
05.14
高效、精準、直觀、實時——取經“蛋白互作研究翹楚”BIFC!
-
05.14
轉染效率低、干擾效果差、重復性欠佳...siRNA研究頻遇“攔路虎”怎么辦?
-
04.22
一文讀懂EMSA技術核心要點,讓“emsa” 秒變“easy”
 X
X 






