
癌癥基因組結構變異的綜合檢測與分析
信息來源:金開瑞 作者:genecreate 發布時間:2018-11-15 13:58:15
題目:Integrative detection and analysis of structural variation in cancer genomes
期刊:Nature genetics
影響因子:27.125
主要技術:Hi-C、集成光學映射(Irys)、全基因組測序(WGS)
研究背景
結構變異(SVs),包括倒置、刪除、復制陽離子和易位,是大多數癌癥基因組的標志。復發性SVs的發現及其對基因組織和表達的分子效應促進我們對腫瘤發生的認識。許多致癌基因已被確認為復發易位的產物,并為藥物治療特別是造血惡性腫瘤提供了成功的靶點。盡管它很重要,但在癌癥基因組中鑒定SVs仍然具有挑戰性。在這里,作者利用高通量染色體構象捕獲(Hi-C)、集成光學映射和全基因組測序,系統地檢測正常或癌癥樣本中的SVs,意圖探究癌基因組中,SVs對突變驅動因素的影響。
研究內容及結果
1. 檢測腫瘤基因組SV的方法
為了評估對SVs不同檢測方法的能力,作者選取了8個癌細胞系和1個典型正常對照(GM12878)(見表1),對它們的WGS、光學測圖和Hi-C數據進行比較(圖1a),發現三種方法均檢測到Caki2細胞中染色體2和3的易位(圖1b),通過觀察同一區域DNA復制時間譜的顯著變化,也證實了這種易位。同時觀察到,與正常細胞相比,癌癥基因組顯示出更多的重組事件,如圖1c所示環狀基因組結構剖面。
表1腫瘤和正常細胞系的高置信度的SVs數
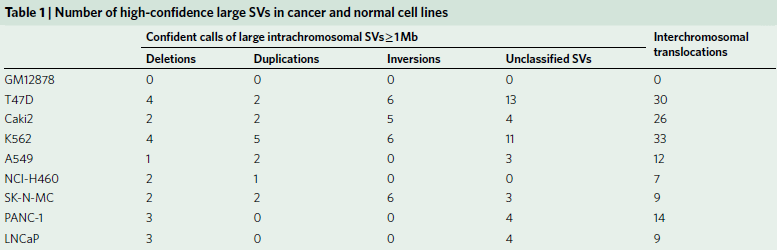
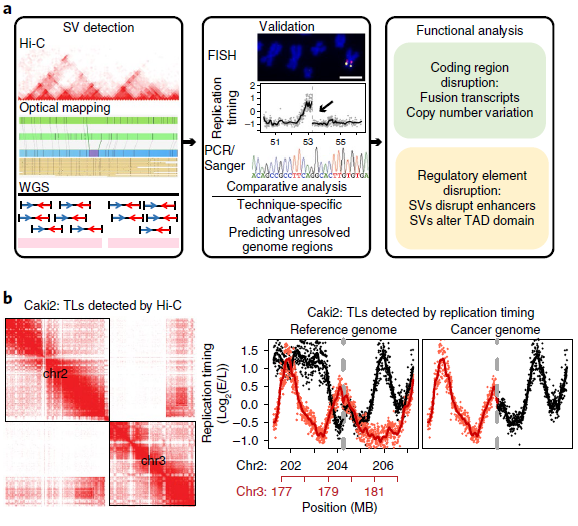
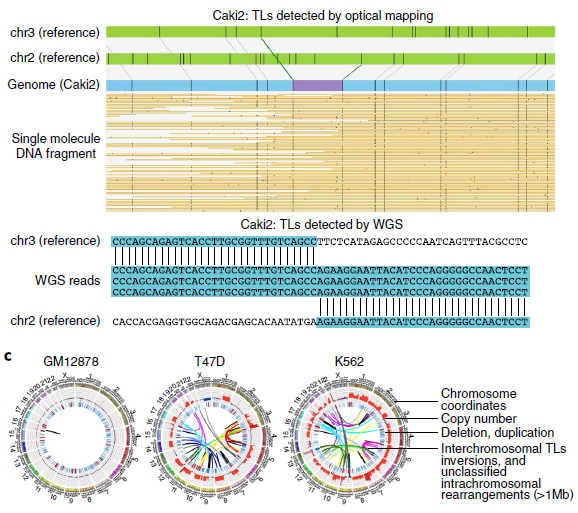
圖1 腫瘤基因組SV檢測的總體策略
2. 利用Hi-C數據檢測大規模重排
在Hi-C實驗中,正常細胞染色體間相互作用非常罕見(圖2a左)。然而,這這種情況在癌細胞中卻相反。例如Caki2癌癥在細胞中,觀察到了強烈的染色體間相互作用(圖2a右),這可能是由于6號染色體和8號染色體的融合。但是關于癌細胞的染色體相互作用增加的信號是由于重組還是三維基因組組織的變異導致的還不清楚,因此針對這一問題,作者首先為“正常”的三維基因組組織特征建立了概率模型,包括位點、TADs、A/B compartments之間的基因組距離,發現小染色體和次端粒區域之間相互作用的增加。并且在重排的情況下,兩個重排區域的基因是融合的,因其改變了位點之間的線性距離,從而也導致了與局部預期交互頻率的偏差(圖2a、b)。
其次,作者利用Hi-C數據進行全基因組SVs檢測,這一檢測屬于一種新型算法。該算法具體體現為:作者首先用一個特征良好的慢性粒細胞白血病細胞系(K562)來評估,并將結果與已發表的核型進行比較。在19個Hi-C預測的重排中,11個可以確認,其余8個是新的。由于這8個均在兩個獨立實驗室進行的,它們不太可能是克隆進化的產物。隨后,作者進行了FISH實驗來驗證新的預測易位。使用Hi-C數據預測的19個易位中有18個通過FISH或以前的核型驗證,結果表明,新算法能夠識別具有高特異性的大規模結構變異。
其次,作者利用Hi-C數據進行全基因組SVs檢測,這一檢測屬于一種新型算法。該算法具體體現為:作者首先用一個特征良好的慢性粒細胞白血病細胞系(K562)來評估,并將結果與已發表的核型進行比較。在19個Hi-C預測的重排中,11個可以確認,其余8個是新的。由于這8個均在兩個獨立實驗室進行的,它們不太可能是克隆進化的產物。隨后,作者進行了FISH實驗來驗證新的預測易位。使用Hi-C數據預測的19個易位中有18個通過FISH或以前的核型驗證,結果表明,新算法能夠識別具有高特異性的大規模結構變異。
最后,將Hi-C分析擴展到27個癌細胞系和9個核型正常細胞系(圖2d),發現在癌細胞中報告了25次重排,在正常細胞中幾乎沒有發生這種情況,染色體間和染色體內重排的比率約為2:1(所有細胞系中為424比274)。因此,新算法似乎可以識別大部分的大SVs,只有4.3%的無法識別。
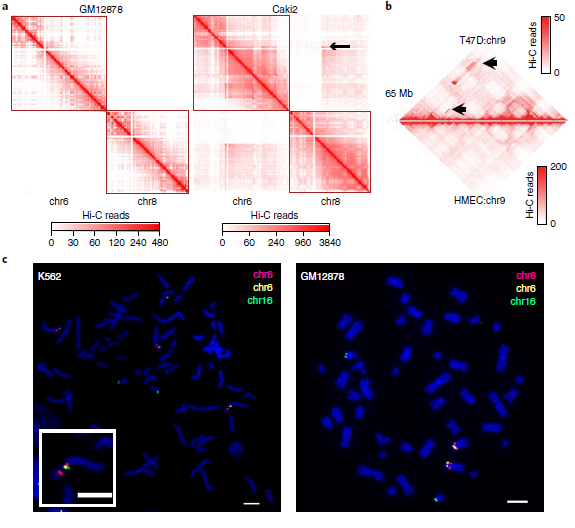
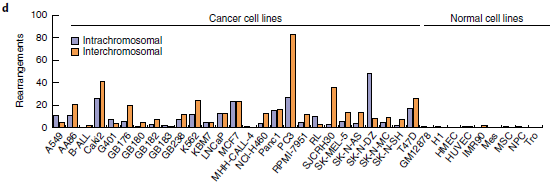
圖2 利用Hi-C數據檢測大規模重排
3. 不同方法檢測SVs的比較
通過光學映射和WGS在每個癌細胞株中鑒定了數千個遺傳物質的增加或損耗,光學映射檢測到的缺失比WGS更少但范圍更大。在T47D細胞中,WGS檢測到2943個缺失,中位大小為552 bp,而Irys檢測到1128個缺失,中位大小為1 335 bp(圖3a,b)。其中85% WGS檢測到的缺失被Irys遺漏,且其中78%的中位大小小于1kb。由于其分辨率受兩個刻痕點之間的最小距離的限制,這些特征很可能被光學映射所忽略。Irys預測的缺失中有3%與多個較小的WGS缺失重疊,在這些情況下,這些WGS缺失的總和大小接近Irys檢測到的缺失,但在Irys檢測到的缺失中,有15%沒有被WGS捕獲。
作者測試了Irys檢測到的一部分缺失,其中87.5%的缺失(16個缺失中的14個)通過了PCR驗證。光學映射可以識別WGS reads沒有被映射的重復區域內的缺失(圖3c),以及在斷點周圍可映射性較低的區域。同時還發現WGS、Irys和Hi-C可以檢測到不同染色體間大規模重排,類似基因組的非模板化添加堿基或外源DNA序列,如病毒的堿基,它可能來自第三條染色體因其太短而無法識別。如圖3d所示,光學映射到局部結構,WGS用來確定斷點,WGS通過定位斷點和Hi-C數據來驗證同一等位基因上幾個相鄰重排。總之,采用互補技術的綜合方法對于更全面地了解癌癥基因組的結構變化至關重要。
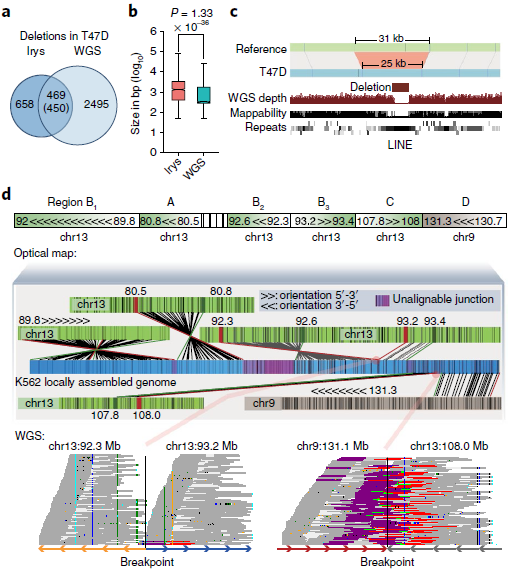
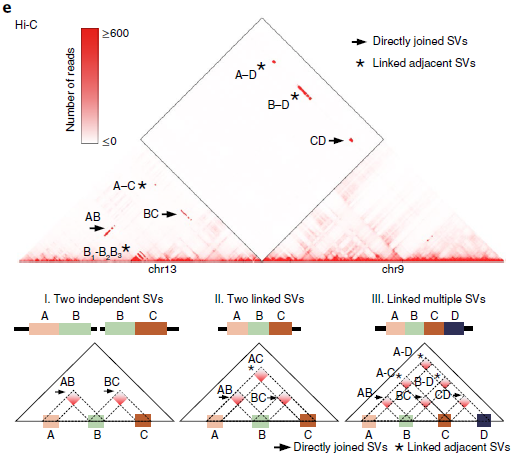
圖3 不同方法檢測SVs的比較
4. SVs對增強子的影響
拷貝數改變(CNAs)代表癌癥的另一類遺傳變異。作者在T47D乳腺癌細胞系中對CNAs進行了分析,并與560例乳腺癌患者的WGS數據進行了比較。在10個最常見的突變癌基因中,有8個在T47D癌細胞中被擴增,ATRX、CDKN1B等腫瘤抑制基因表達缺失(圖4a),說明T47D細胞反映了乳腺癌的CNA表達情況。
作者進一步比較了T47D和人乳腺上皮細胞(HMECs)的RNA-seq數據,發現雜合性(LOH)缺失和純合缺失導致基因表達顯著降低,并且在其他癌細胞株中也觀察到這一點。作者在25個COSMIC(癌癥體細胞突變目錄)發現與腫瘤相關的基因,大多數(76%)顯示轉錄降低。而已知的癌基因(如MYC)和細胞周期檢查點基因(如CDKN2A和CDKN2B)廣泛擴增。
為了研究SVs是否可以通過破壞遠端調控元件來影響癌癥相關基因的表達。作者重點比較了T47D乳腺癌細胞與人乳腺上皮細胞(HMECs)。主要方法是使用ENCODE 聯合 (URLs)的H3K27ac染色質免疫沉淀測序數據預測HMECs中的增強子,并將增強子與T47D中的缺失區域進行比較。結果表明,GNB4基因下游的3.4 kb缺失與乳腺組織特異性增強子重疊。由于基因組擴增,該區域有6個拷貝,其中5個帶有這種缺失,只有單拷貝的增強子沒有被破壞。
HMECs中的Hi-C數據表明GNB4可能受到單拷貝增強子的調控。更重要的是,它是該區域唯一表達減少的基因,該區域其余基因的表達高度上調,可能是由于拷貝數增加(圖4c)。此外,發現缺失的增強子位于乳腺癌相關通路的基因附近(圖4d),并且連接這些缺失的增強子相關的基因表達水平降低(圖4e)。總的來說,這些結果表明癌癥基因組的缺失可能經常影響增強子,并可能促進腫瘤發生。
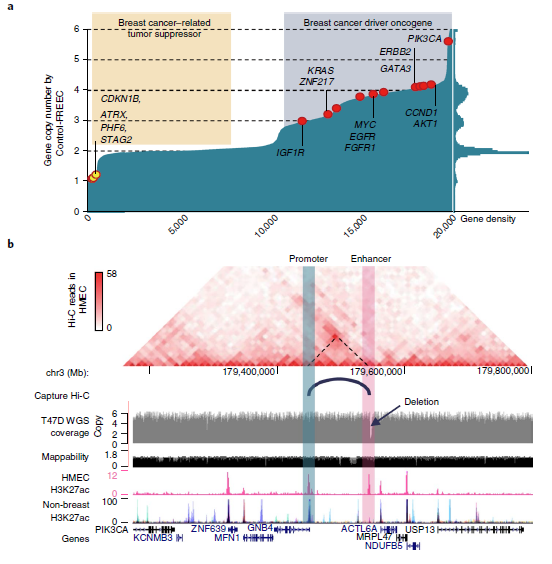
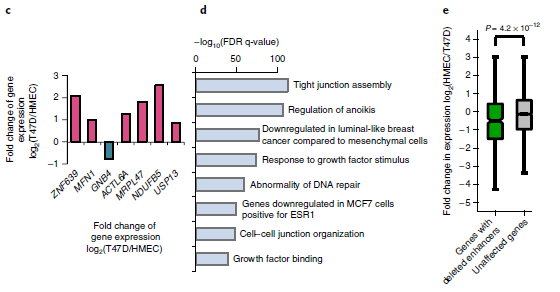
圖4 SVs對增強子的影響
5. 結構變異對三維基因組組織的影響
基因突變會破壞拓撲結構域(TADs)并產生“新TADs(neo-TADs)”,導致發育障礙中的基因表達失調。為了研究SVs對三維基因組組織的影響,作者利用Hi-C數據鑒定了20個癌細胞株中的SVs,系統地研究了結構變化對TAD結構的影響,觀察到neo-TADs是癌細胞大規模基因組重排的結果。如圖5a所示,在PANC-1細胞中,染色體9和18的融合形成了一個neo-TAD。此外,發現許多由SVs誘導的neo-TADs在癌細胞中含有已知癌癥驅動基因,如ERBB2、ETV1、ETV4、MYC、TERT等。
為了探討neo-TAD形成是否是癌癥基因組SV重排的一般結果,作者對每個細胞系中的所有斷點交叉Hi-C信號進行了匯總分析。如圖5b所示,觀察到染色體間Hi-C信號形成一個尖銳的三角形(虛線),表明由于重排而形成的融合TAD。當使用隨機邊界位置打亂TADs進行同樣的分析時,沒有觀察到這種情況。這些結果表明,癌癥中的結構變異可以重組TAD結構,導致TAD融合和調節環境的改變(圖5c)。
接著,作者研究了neo-TADs對基因表達的影響。在8個癌細胞系中,觀察到包含重排的TADs基因比未重排的TADs基因表現出更大的等位基因偏差,這表明neo-TADs可能導致基因表達的改變。接著,作者檢查了3個神經母細胞瘤細胞系的Hi-C數據,并比較了MYC的表達。其中SK-N-DZ具有較高的MYCN/N-myc表達(圖5e),其余兩個SK-N-SH和SK-N-AS具有較高的MYC/c-Myc表達。值得注意的是,在兩個高MYC表達的神經母細胞瘤細胞系(SK-N-SH和SK-N-AS)中,發現了MYC基因附近存在易位。據癌癥細胞系百科全書數據記載,這兩個細胞系中沒有MYC擴增,而作者觀察了兩種情況下包含MYC基因的neo-TADs的形成(圖5f,g),表明neo-TADs的形成可能與MYC激活有關。綜上所述,neo-TADs的產生是癌癥基因組重組的結果。

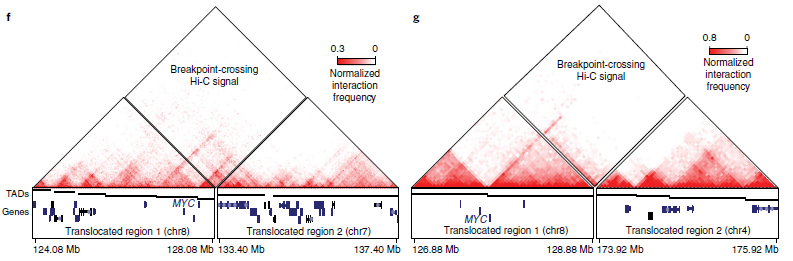
圖5 重排和TAD融合
文章小結
作者利用高通量染色體構象捕獲(Hi-C)、集成光學映射和全基因組測序,系統地檢測正常或癌癥樣本中的SVs,發現光學映射和Hi-C能有效檢測大而復雜的結構變化,高覆蓋的WGS能識別高分辨率SVs。此外,還發現結構基因組變異而導致的三維(3D)基因組組織改變的實例,例如拓撲關聯域(TADs)的形成或分解,這表明結構變異在腫瘤發生的基因失調中起著關鍵作用。
解析文獻
Jesse R. Dixon, Jie Xu ,et al. Integrative detection and analysis of structural variation in cancer genomes. Nature Genetics, 2018, 50: 1388–1398.
參考文獻
1. Northcott, P. A. et al. Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma. Nature ,2014, 511, 428–434.
2. Dileep, V. et al. Topologically associating domains and their long-range contacts are established during early G1 coincident with the establishment of the replication-timing program. Genome Res. 2015, 25, 1104–1113.
3. Haas, B. et al. STAR-Fusion: fast and accurate fusion transcript detection from RNA-Seq. Preprint at https://www.biorxiv.org/content/ early/2017/03/24/120295,2017.
4. Marchal, C. et al. Genome-wide analysis of replication timing by nextgeneration sequencing with E/L Repli-seq. Nat. Protoc.2018, 13, 819–839.
最新動態
-
09.23
中藥的現代詮釋:外泌體如何革新傳統醫學?
-
07.02
1+1>2!深度解析RNA測序數據挖掘邏輯和后期實驗設計思路,輕松研獲10+ SCI
-
07.01
“稻”亦有道——盤點近期水稻研究的重大突破
-
06.28
科學與美學的結合體:植物亞細胞定位技術詳解
-
06.28
“聚焦新質生產力,激發科研新動能”|LCA躋身蛋白互作研究的新銳力量
-
06.05
知無不“研”|一文讀懂免疫共沉淀技術(Co-IP)
-
05.14
四大研究利器(Co-IP、BIFC、Y2H、GST pull-down)助力速配蛋白互作“最佳拍檔”
-
05.14
高效、精準、直觀、實時——取經“蛋白互作研究翹楚”BIFC!
-
05.14
轉染效率低、干擾效果差、重復性欠佳...siRNA研究頻遇“攔路虎”怎么辦?
-
04.22
一文讀懂EMSA技術核心要點,讓“emsa” 秒變“easy”
 X
X 






