
【詳解】基于圖像的單細胞空間分辨轉錄組學技術
單細胞多組學技術被Nature Methods評為2019年年度技術方法,緊接著空間轉錄組就被Nature Methods評為2020年年度技術方法,單細胞轉錄組測序加空間轉錄組聯合分析已經是目前炙手可熱的轉錄組研究方法,是研究細胞異質性、組織形態學、組織發育機制和細胞圖譜等領域的不二選擇。
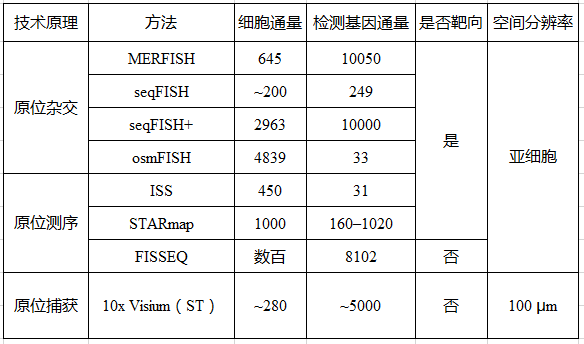
目前廣泛應用的空間轉錄組技術10x visium(收購了Spatial transcriptomics技術),原理為原位捕獲(in situ capturing),其不足之處是芯片一個位點上有1~10個細胞,無法做到單細胞級分辨率,且無法檢測低表達量基因。MERFISH(multiplexed error-robust fluorescence in situ hybridization,多路抗錯熒光原位雜交)發明者莊小威在Nature Methods發表了Spatially resolved single-cell genomics and transcriptomics by imaging一文,介紹了基于成像原理實現單細胞空間分辨轉錄組學的兩類技術(表 1),這些技術能將RNA定位在亞細胞位置。
熒光原位雜交(Fluorescence in situ hybridization, FISH)應用熒光標記探針定位核酸分子,單分子水平FISH(smFISH)能夠高度精確定位RNA在細胞中的位置。然而,有限數量的顏色通道限制了FISH的通量,只能檢測10~30種RNA。為解決這個問題,2013年莊小威團隊開發了MERFISH,通過來自多輪雜交的信號組合來識別基因,每個基因都分配一個N位二進制條形碼,每位的“1”或“0”值對應于該基因是否在一個特定輪的雜交中被檢測到(圖1a)。該二進制編碼方案可以通過N輪雜交區分2N個基因(當在N輪雜交中使用C種顏色進行成像可達到2NC個基因)。現在MERFISH可以通過23輪雜交和3色成像,實現了在單細胞中對大于10,000個基因成像。
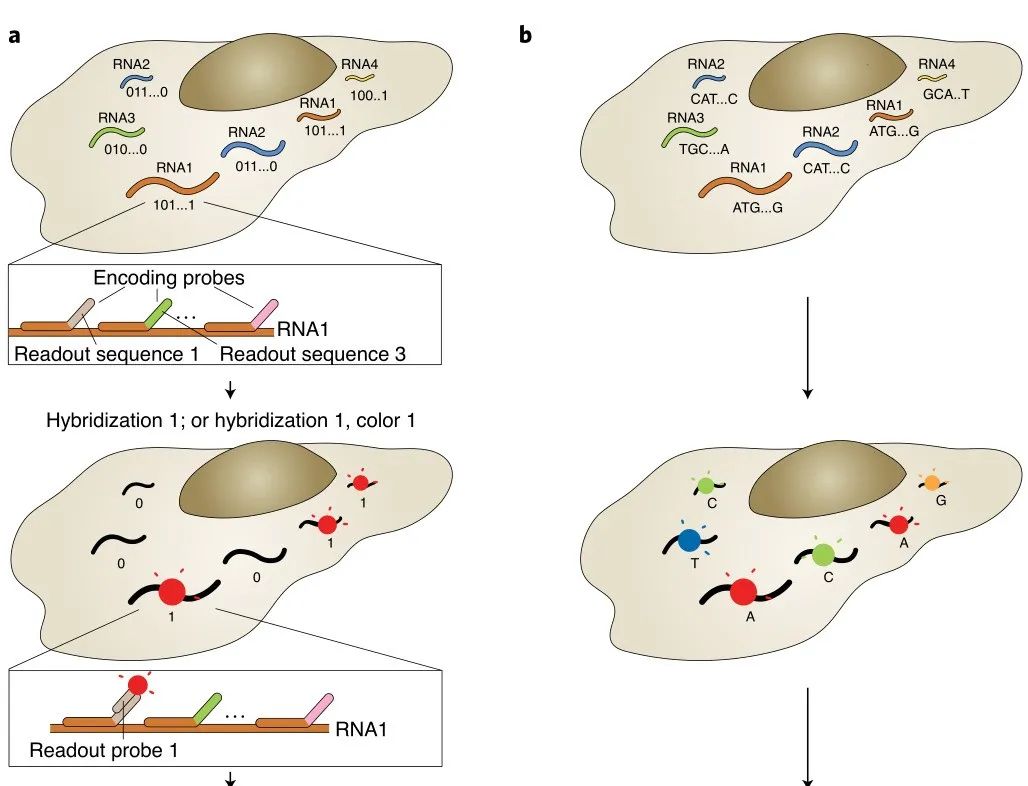
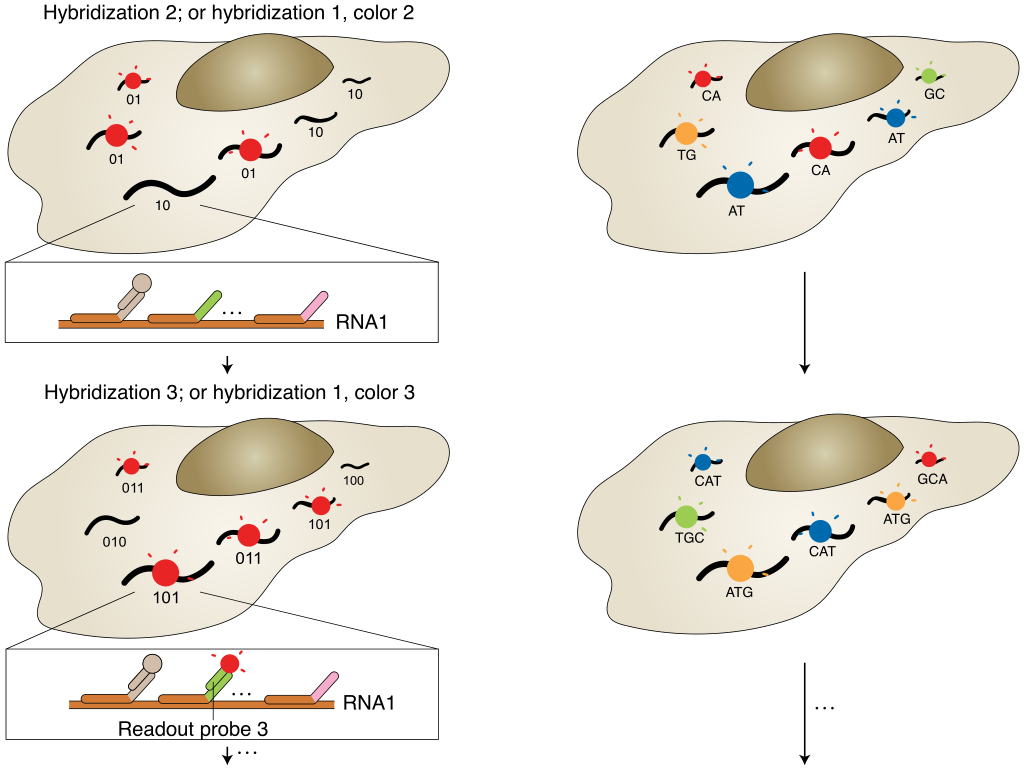
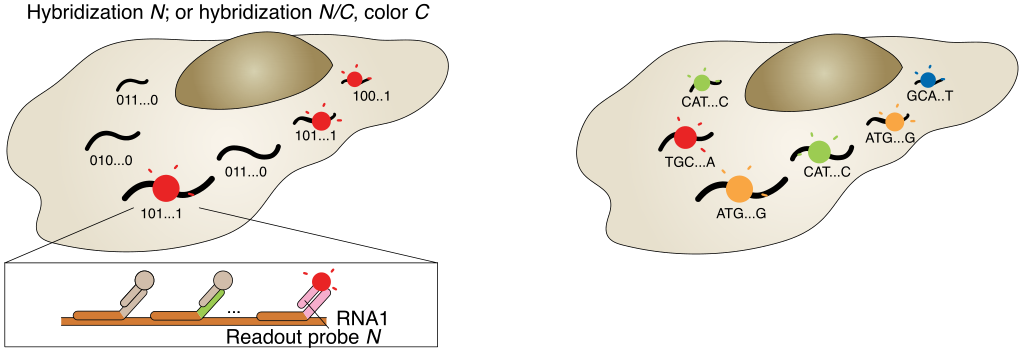
a,以MERFISH方法為例的多路FISH方案。本文展示了一種二進制條形碼方案,以及條形碼如何用編碼探針庫印跡在RNA上,并使用不同的讀出探針逐位檢測。b,原位測序方案示意圖。核酸序列可以是基因的內源性序列,也可以是分配給基因的條形碼序列,通過測序進行逐個核苷酸檢測,圓圈的顏色可能表示單個核苷酸或相鄰的二核苷酸對。
另一種多路FISH方法seqFISH,通過順序顏色編碼來區分RNA,在seqFISH中,不同的RNA組合在每一輪雜交中被分配不同的顏色,之后用一組新的顏色組合的FISH探針與細胞RNA雜交,通過C色和N輪雜交,可以區分CN不同的基因。最初seqFISH實現了單細胞中12個基因的定位,之后通過引入雜交鏈信號放大實現了約250個基因成像。然而,顯微成像時分子顏色堆疊問題限制了seqFISH的通量發展。
空間分辨的單細胞分析也通過序列雜交實現,例如osmFISH在每個雜交的每個顏色通道中對應單個基因。這種方法在組織成像中提供了極好的信號質量,盡管其多路復用性水平較低,但提供了一種對表達水平過高或序列過短的基因進行成像的方法,它們通常無法進行單分子檢測或條形碼識別。這種方法也可以與上述方法相結合,可以覆蓋多數的基因和難以進行單分子檢測的基因。
總的來說,多路FISH方法提供了具有高空間分辨率的原位單細胞轉錄組學和3D基因組分析。這些方法的另一個優點是檢測效率高。MERFISH已經證明了對超過10000個基因中的80%成像,如此高的檢測效率可以定量分析低表達水平的基因。
與此同時,基于成像的原位測序方法也被開發出來,用于具有高空間分辨率的單細胞轉錄組分析,在細胞中對RNA進行原位測序,四種顏色熒光各代表一種核苷酸,根據熒光信號記錄序列信息(圖1b)。不同于原位雜交針對靶向RNA,原位測序能以靶向或非靶向的方式進行。
在最初原位測序(ISS)技術中,需在靶向原位測序中預選一組基因,將特異性核苷酸序列作為條形碼通過掛鎖探針雜交傳遞到這些基因,然后進行循環擴增和原位測序檢測。STARmap使用條形碼掛鎖探針,與靶標雜交,通過添加第二個引物,針對掛鎖探針旁邊的位點,避免了逆轉錄(RT)步驟,并降低了噪音。STARmap能夠對完整的組織樣本進行3D分析,保留了細胞的方向性,而不僅僅是單一的2D層。但需要注意的是,這種能力只表現在100~150微米厚的切片和較少的目標數量上。STARmap能夠對單細胞中1020個目標基因成像,檢測效率與單細胞RNA測序相當。
在非靶向原位測序中,例如FISSEQ(Fluorescence in situ sequencing,熒光原位測序),引入了細胞RNA逆轉錄為cDNA后擴增,并無需任何預選靶向序列。這種方法的優點是能做到全基因組覆蓋,FISSEQ已經在同一樣本中對大于8000個基因成像。然而,由于所有種類的RNA都被測序,為了避免分子擁擠的問題,FISSEQ需要使用測序引物檢測單個分子,導致只有一小部分的擴增子被測序,使得檢測效率較低,為全基因組0.2%到<0.01%。
對于所有的靶向方法,包括原位測序和多路FISH,這是克服分子堆疊以及提高檢測范圍一種簡單的方法——將目標基因分成多個組,一次一組成像。例如,10000或20000個基因可以分為10或20組1000基因(通常不超過10輪多色成像),對10000或20000基因成像不需要改變方法,只是進行多次10或20輪成像。
之后小編將帶來基于圖像的單細胞空間分辨和轉錄組學技術應用介紹,敬請期待!
1.Method of the Year 2019: Single-cell multimodal omics. Nat Methods. 2020;17(1):1. doi:10.1038/s41592-019-0703-5
2.Method of the Year 2020: spatially resolved transcriptomics. Nat Methods. 2021;18(1):1. doi:10.1038/s41592-020-01042-x
3.Ståhl PL, Salmén F, Vickovic S, et al. Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science. 2016;353(6294):78-82. doi:10.1126/science.aaf2403
4.Zhuang X. Spatially resolved single-cell genomics and transcriptomics by imaging. Nat Methods. 2021;18(1):18-22. doi:10.1038/s41592-020-01037-8
5.Liao J, Lu X, Shao X, Zhu L, Fan X. Uncovering an Organ's Molecular Architecture at Single-Cell Resolution by Spatially Resolved Transcriptomics. Trends Biotechnol. 2021;39(1):43-58. doi:10.1016/j.tibtech.2020.05.006
最新動態
-
09.23
中藥的現代詮釋:外泌體如何革新傳統醫學?
-
07.02
1+1>2!深度解析RNA測序數據挖掘邏輯和后期實驗設計思路,輕松研獲10+ SCI
-
07.01
“稻”亦有道——盤點近期水稻研究的重大突破
-
06.28
科學與美學的結合體:植物亞細胞定位技術詳解
-
06.28
“聚焦新質生產力,激發科研新動能”|LCA躋身蛋白互作研究的新銳力量
-
06.05
知無不“研”|一文讀懂免疫共沉淀技術(Co-IP)
-
05.14
四大研究利器(Co-IP、BIFC、Y2H、GST pull-down)助力速配蛋白互作“最佳拍檔”
-
05.14
高效、精準、直觀、實時——取經“蛋白互作研究翹楚”BIFC!
-
05.14
轉染效率低、干擾效果差、重復性欠佳...siRNA研究頻遇“攔路虎”怎么辦?
-
04.22
一文讀懂EMSA技術核心要點,讓“emsa” 秒變“easy”
 X
X 






