
文獻解讀 | 高密度脂蛋白通過miR-181a-5p調控自噬影響血管新生
正常的高密度脂蛋白(nHDL)在防治組織缺血性疾病中起到積極作用,促進血管生成。相反,在冠心病等病理情況下,高密度脂蛋白(HDL)的功能可能受損,失去促進血管生成的效果,即失功能HDL(dHDL)。自噬是維持細胞和生物體穩態的重要機制。微小RNA(miRNA)在調節血管生成中發揮關鍵作用。目前尚不清楚HDL是否通過miRNA調節自噬來影響血管新生,以及dHDL是否在調節自噬方面與nHDL有所不同。
近日,中山大學附屬第一醫院區景松和歐志君課題組在Science China Life Sciences發表了題為“High-density lipoprotein regulates angiogenesisby affecting autophagy via miRNA-181a-5p”的研究論文,揭示了HDL通過miR-181a-5p調控自噬流影響血管新生的新機制。

nHDL通過刺激自噬和eNOS表達增加一氧化氮(NO)生成,從而促進血管生成。相反,dHDL通過增加miR-181a-5p表達降低自噬和eNOS表達,導致NO產生減少,最終抑制血管生成。這一發現提供了高密度脂蛋白調節血管生成的新機制,為治療dHDL妨礙的血管生成提供了治療靶點。
技術路線
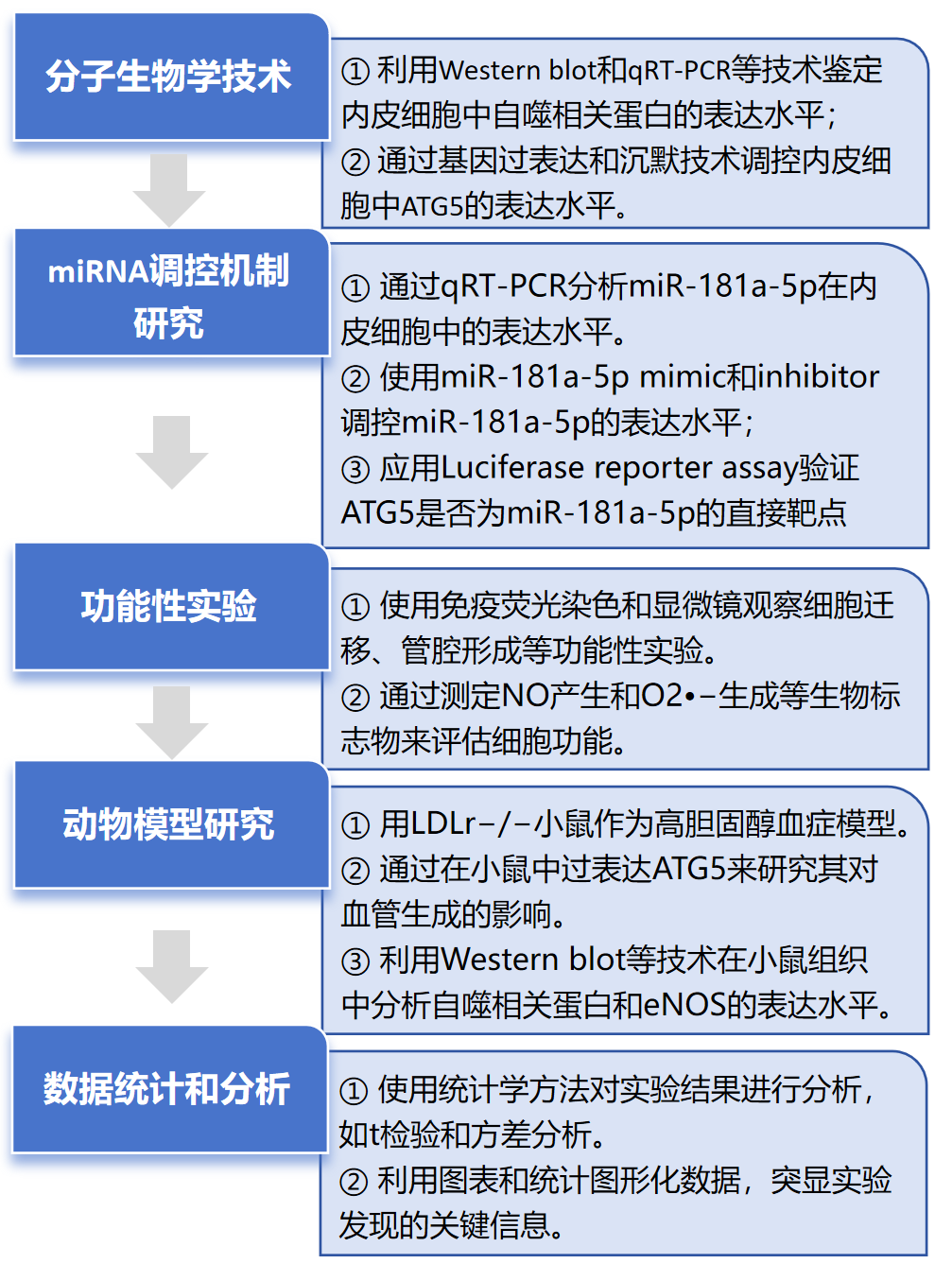
其中Luciferase reporter assay由金開瑞生物合作完成
研究方法及結果
nHDL促進HUVECs中的自噬,而dHDL抑制
研究使用了人臍靜脈內皮細胞(HUVECs)來調查正常高密度脂蛋白(nHDL)和冠心病患者高密度脂蛋白(dHDL)對自噬的影響。通過測量自噬相關蛋白和使用熒光探針等方法,研究發現,與nHDL相比,dHDL顯著增加了細胞內SQSTM1/P62水平,減少了LC3-II的表達。此外,nHDL促進了ATG5、Beclin1和PI3K-CIII等自噬相關蛋白的表達,而dHDL則產生相反的效應。通過使用熒光標記系統,研究還觀察到nHDL增加了細胞內自噬體和自溶體的形成,而dHDL則抑制了這一過程。這些結果表明,nHDL和dHDL對HUVECs中的自噬產生不同的調節作用。
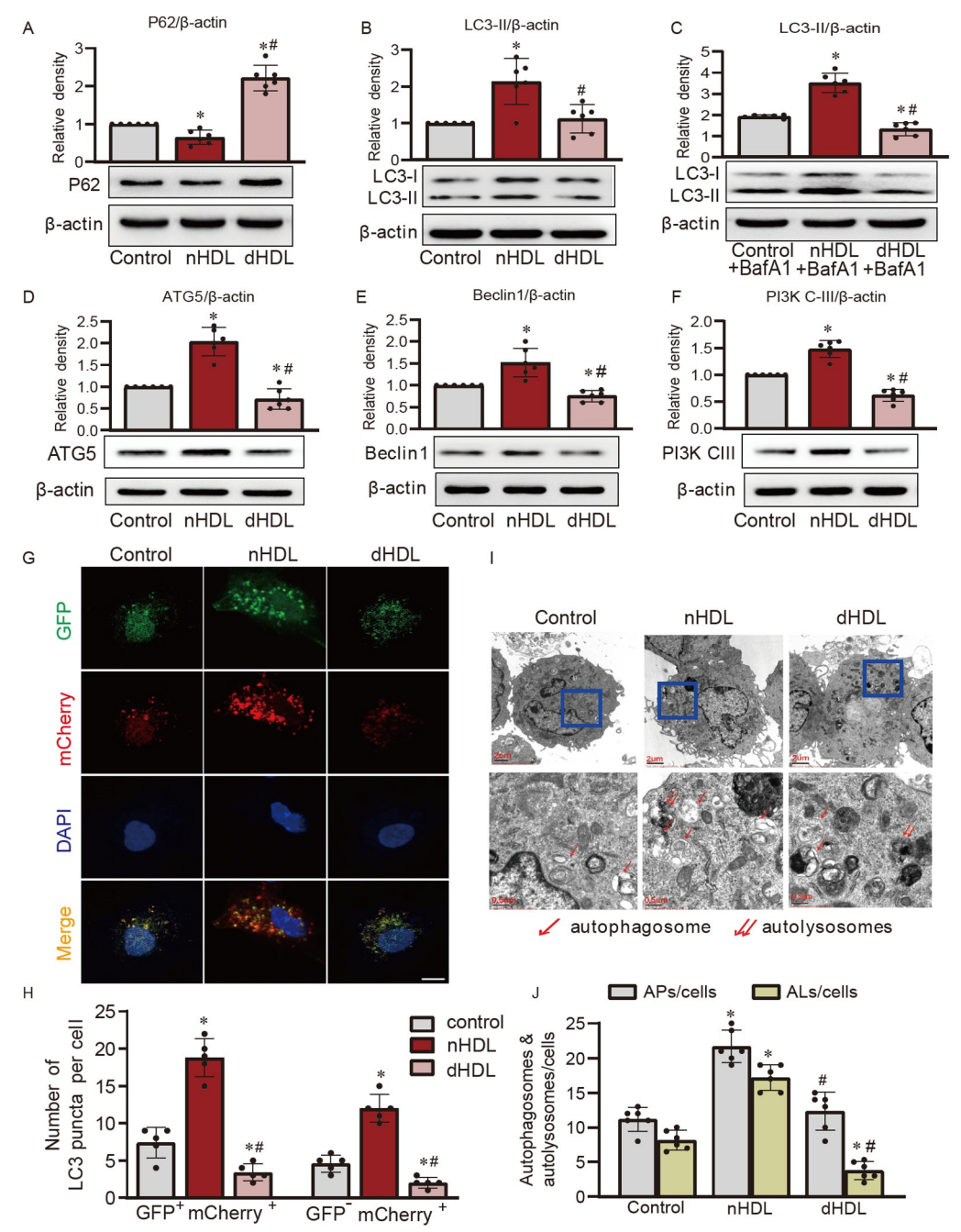
圖1. nHDL和dHDL對內皮細胞自噬的影響。
A–C,Western blot和定量分析顯示在nHDL或dHDL刺激下,HUVECs中LC3-II和P62的水平;D–F,在nHDL或dHDL刺激下,通過Western blot檢測HUVECs中ATG5、Beclin1和PI3K-CIII的表達,;G,通過共聚焦圖像顯示nHDL和dHDL對表達自噬通路報告基因mCherry-GFP-LC3的HUVECs中自噬的影響;H,對在合并圖像中測得的自噬體(GFP+mCherry+)和自溶體(GFP−mCherry+)的定量分析;I和J,電鏡圖像和定量分析顯示在nHDL或dHDL刺激下HUVECs中的自噬體或自溶體。
在人臍靜脈內皮細胞(HUVECs)中通過抑制自噬降低eNOS和NO的產生
通過使用3-甲基腺嘌呤(3-MA)抑制自噬,驗證了nHDL和dHDL誘導的自噬在內皮細胞(ECs)中的不同功能。圖2A和B顯示,當抑制自噬時,nHDL導致的PI3K-CIII和eNOS的上調以及P62的下調被抑制。抑制自噬時,nHDL誘導的NO產生受阻(圖2C–E)。自噬抑制增加了O2•−的產生(圖2F和G)。這些數據為我們了解nHDL如何通過自噬途徑保護ECs免受氧化應激提供了新的見解。
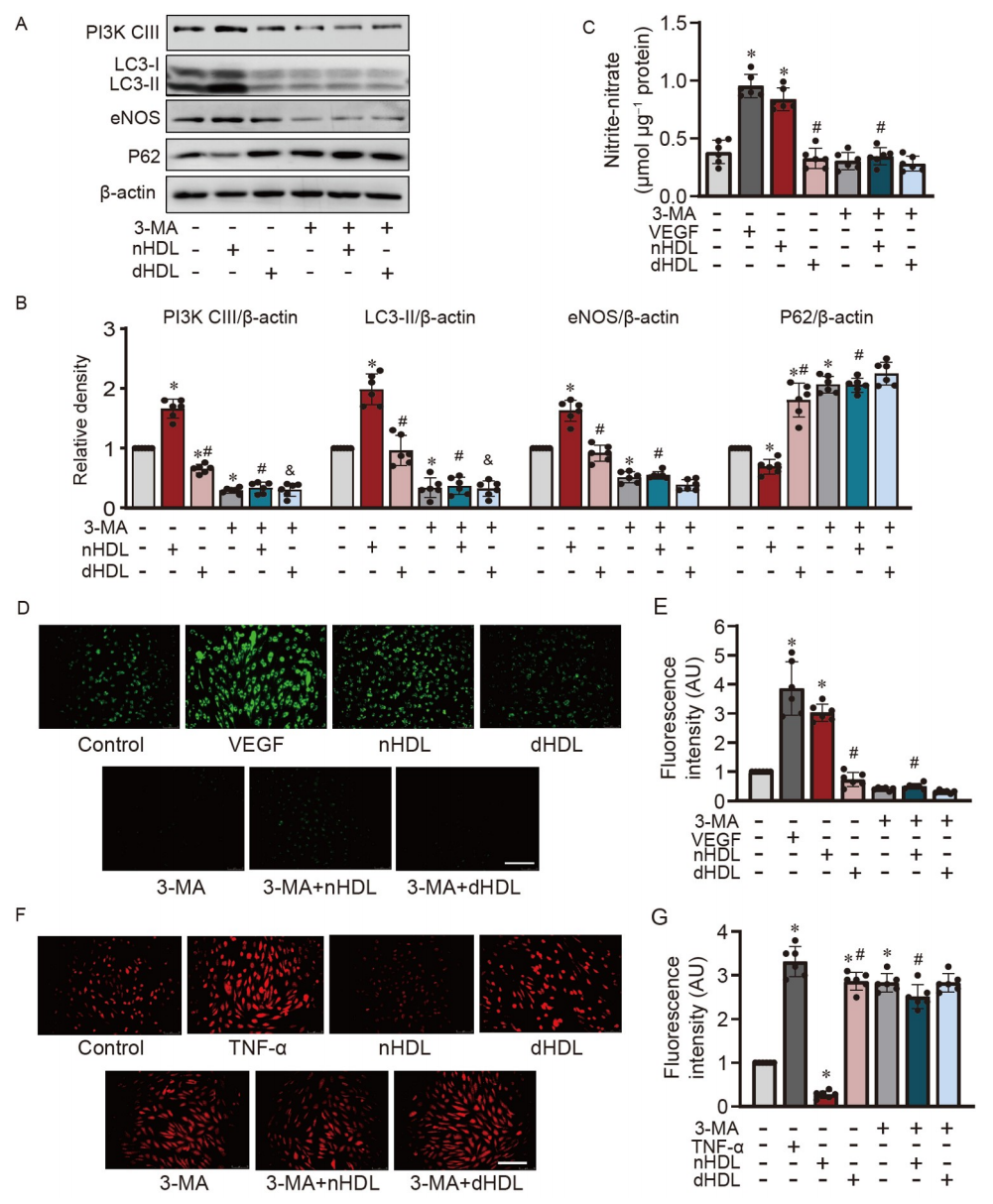
圖2. 抑制自噬對nHDL促進HUVECs中eNOS表達、NO產生和O2•−生成的影響。
A,Western blot檢測經nHDL或dHDL處理的HUVECs中LC3-I/II、eNOS、PI3K-CIII和P62的表達;B,蛋白定量相對于beta-actin表達;C,使用Sievers NOA分析儀顯示HUVECs中的NO產生的條形圖;D和E,使用DAF-2DA檢測在HUVECs中經nHDL或dHDL處理后細胞內NO的圖像和定量;F和G,在HUVECs中,圖像和定量顯示3-MA通過抑制自噬增強了對照組和nHDL處理組中內皮O2•−的生成。
通過沉默ATG5抑制自噬導致內皮細胞中O2•−的增加,并減弱了nHDL誘導的內皮細胞遷移和血管形成
ATG5在自噬體形成中至關重要,對于細胞分化和穩態維持起著重要作用。由于nHDL和dHDL對ATG5有顯著不同的影響(圖1D),作者選擇ATG5作為自噬干預的靶點。在HDL處理之前沉默了ATG5的表達。ATG5的沉默效果顯示在支持信息的圖S2中。ATG5的沉默降低了LC3-II和eNOS的表達(圖3A和B),增加了P62的表達,阻斷了nHDL誘導的eNOS表達和NO產生(圖3C和G),增強了O2•−的生成(圖3D和H)。此外,沉默ATG5抑制了血管內皮細胞nHDL誘導的細胞遷移(圖3E和I)和管形成(圖3F和J)。
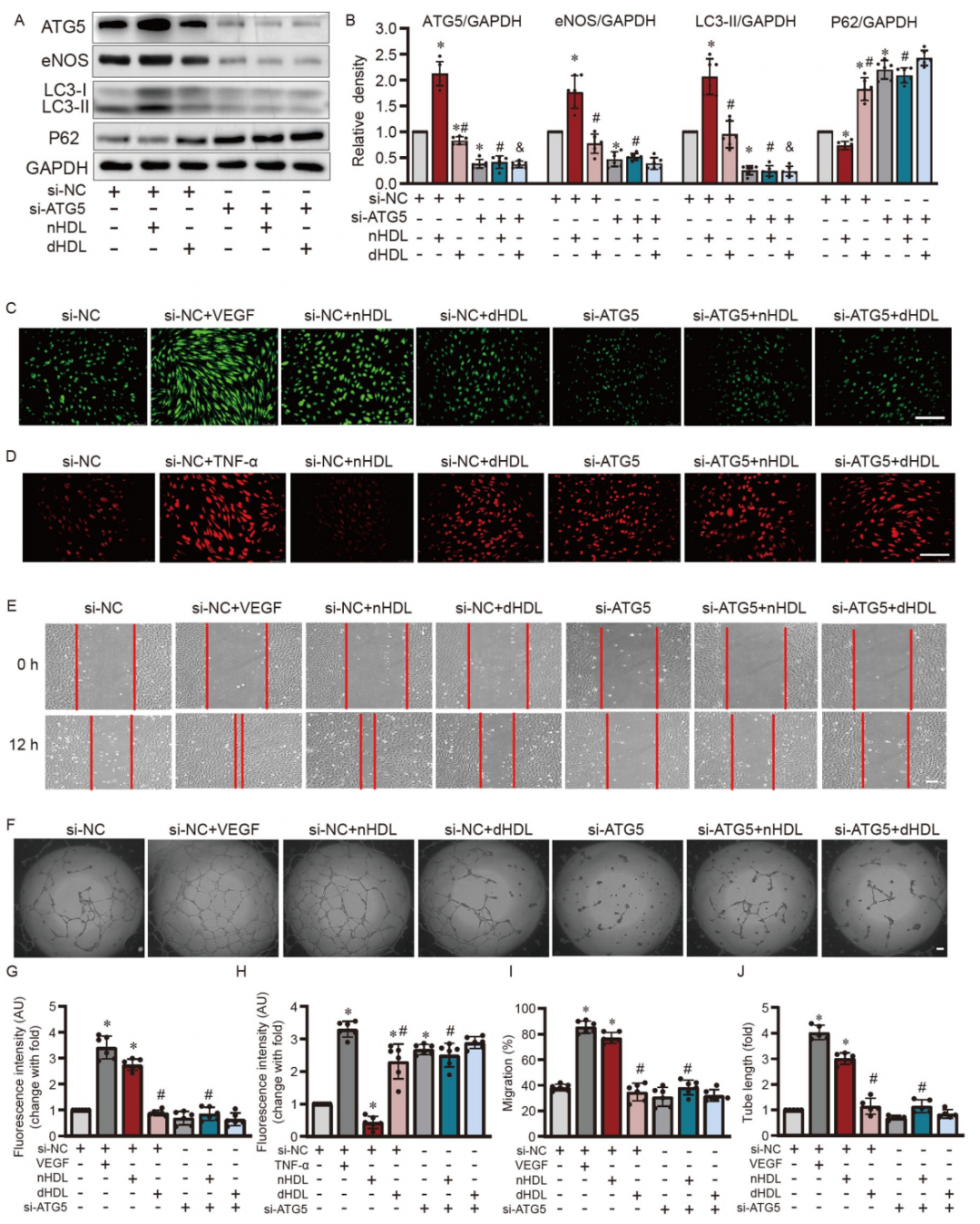
圖3. ATG5沉默和nHDL誘導的eNOS表達、NO產生、細胞遷移、管形成、以及在HUVECs中O2•−生成的定量分析。
A和B,Western blot和定量分析顯示ATG5的沉默降低了eNOS和LC3-II的表達,同時增加了P62在培養的HUVECs中的表達;C和D,圖像顯示當通過ATG5沉默減弱自噬時,nHDL或dHDL對NO產生和O2•−生成的影響;E和F,圖像顯示siRNA沉默ATG5抑制了nHDL誘導的HUVECs中的遷移和血管形成;G–J,定量分析顯示siRNA沉默ATG5抑制了nHDL誘導的HUVECs中的NO產生、遷移、血管形成,以及O2•−生成。
通過過表達ATG5來刺激自噬,挽救了dHDL的血管生成作用
為了探究刺激自噬是否能夠逆轉dHDL的血管生成效應,研究者在內皮細胞中過表達了ATG5。ATG5的過表達增加了LC3-II和eNOS的表達,降低了在dHDL刺激下的P62的表達。ATG5的過表達恢復了由dHDL抑制的NO產生,減少了dHDL刺激下的O2•−生成。重要的是,ATG5的過表達在dHDL刺激下恢復了內皮細胞的遷移和管道形成。這些數據表明ATG5可能是nHDL保護作用的介導者,并對內皮細胞的血管生成產生了顯著影響。
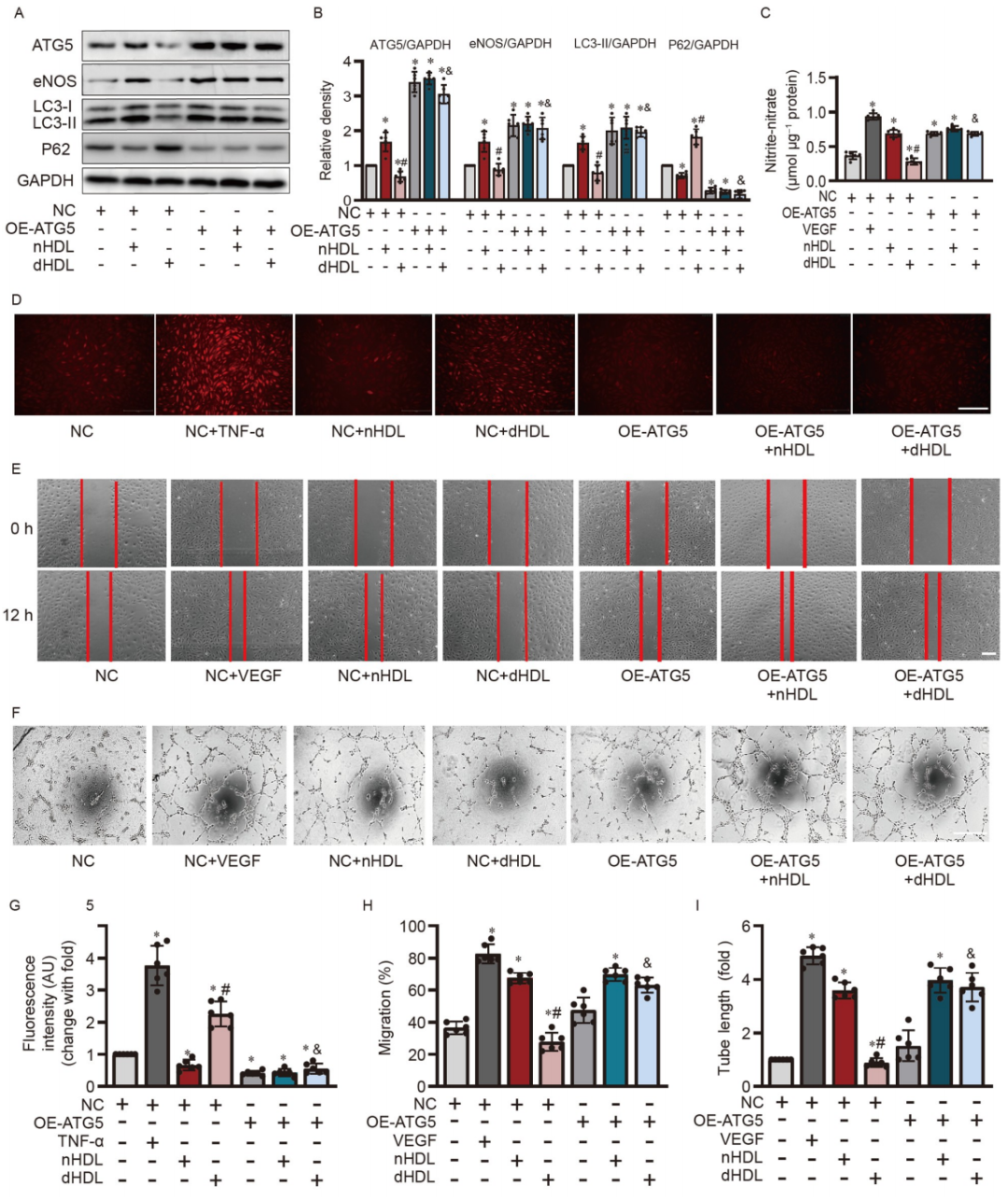
圖 4. ATG5的過表達恢復了dHDL抑制的HUVECs中eNOS的表達,NO的產生,遷移和管道形成,并降低了O2•−的生成。
A和B,Western blot和定量分析顯示ATG5的過表達增加了eNOS和LC3-II的表達,并抑制了P62在培養的HUVECs中的表達;C,檢測了在過表達ATG5后,nHDL或dHDL處理下HUVECs中的內皮NO產生,NO的產生增加,并恢復了dHDL促進HUVECs中NO產生的能力;D,圖像顯示了ATG5的過表達減少了dHDL誘導的HUVECs中O2•−的生成;E和F,圖像顯示了ATG5的過表達,恢復了HUVECs中dHDL誘導的遷移和血管生成。G–I,對D–F圖像的定量分析。
nHDL和dHDL通過miR-181a-5p以不同方式調控ATG5
研究者重新驗證了數據(Li等,2020),證實miR-181a-5p在nHDL作用下降低而在dHDL作用下上升,通過定量實時聚合酶鏈反應(qRT-PCR)進行檢測(圖5A)進一步發現nHDL抑制了pre-miR-181-1和pre-miR-181-2,而dHDL刺激了它們(圖5B)。在沉默SRB1后,nHDL和dHDL對miR-181a-5p的影響被消除(圖5C),表明它們通過SRB1受體在ECs中調節miR-181a-5p的表達。TargetScan(targetscan.org)顯示ATG5可能是miR-181a-5p在HUVECs中的潛在靶點(圖5D)。miR-181a-5p模擬物抑制了ATG5的Luciferase活性,驗證了ATG5是miR-181a-5p的直接靶標(圖5E)。將miR-181a-5p模擬物和抑制物轉染到HUVECs后,miR-181a-5p模擬物降低了ATG5的表達,而miR-181a-5p抑制物增加了它們(圖5F–H),同時eNOS的表達也受到了類似的調控(圖5G和H)。
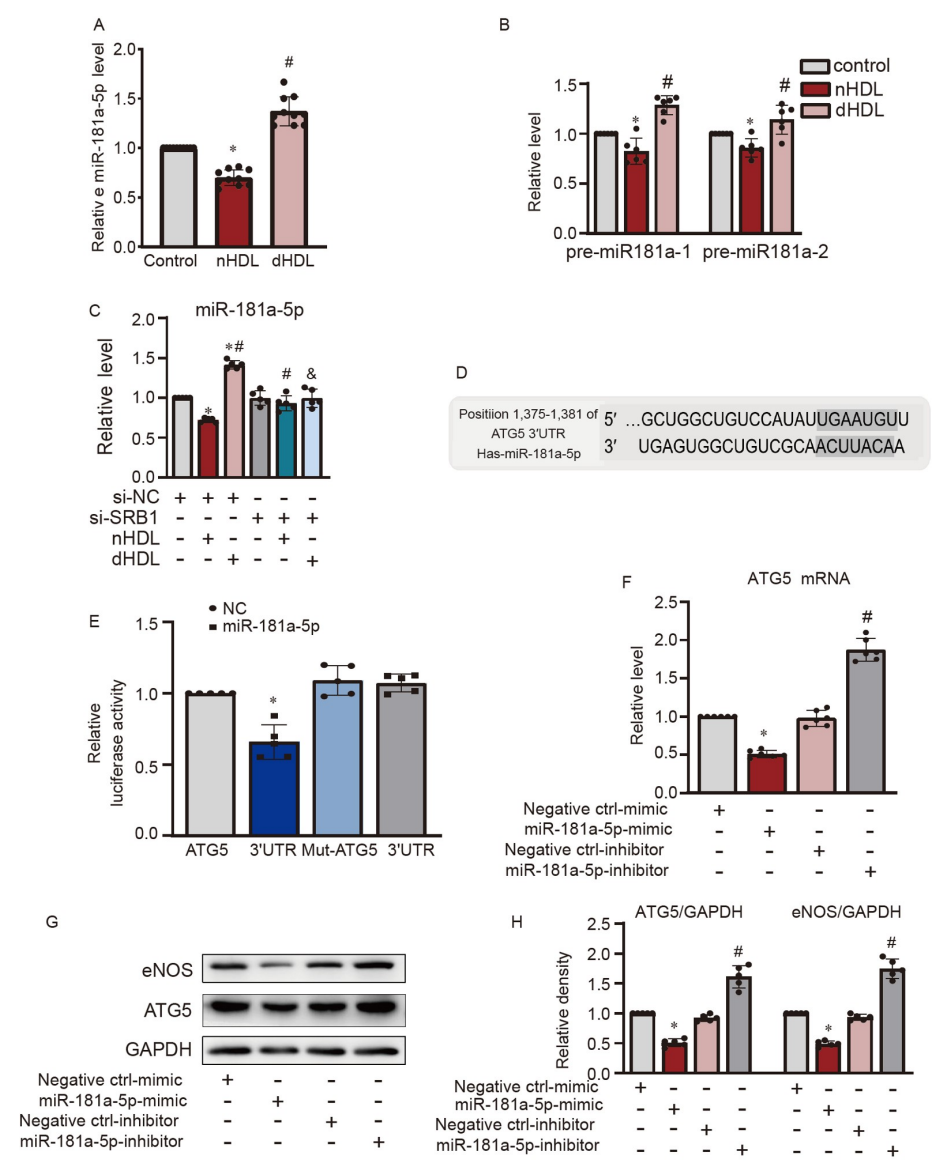
圖5. HDL對內皮miR-181a-5p和ATG5表達的影響
A,qRT-PCR顯示nHDL和dHDL對培養的HUVECs中miR-181a-5p表達的影響;B,qRT-PCR顯示nHDL和dHDL對培養的HUVECs中pre-miR-181-1和pre-miR-181-2表達的影響;C,qRT-PCR顯示在沉默SRB1后,nHDL和dHDL對培養的HUVECs中miR-181a-5p表達的影響;D,ATG5中可能的人類miR-181a-5p結合位點;E,Luciferase活性確認ATG5在HUVECs中是miR-181a-5p的直接靶標;F,miR-181a-5p模擬物降低了HUVECs中ATG5的mRNA表達,而miR-181a-5p抑制物則增加了它;G和H,miR-181a-5p模擬物或miR-181a-5p抑制物轉染到HUVECs中后,eNOS和ATG5的免疫印跡分析的代表圖像和定量分析。
研究者進一步研究了HDL是否通過miR-181a-5p調節自噬。miR-181a-5p模擬物抑制了LC3-II的表達,增加了P62的表達,阻斷了自噬體的合成(圖6A、C、E和F)。miR-181a-5p抑制物增加了LC3-II的表達,抑制了P62,促進了自噬體的合成(圖6B、D、E和G)。miR-181a-5p模擬物抑制了nHDL誘導的自噬,miR-181a-5p抑制物逆轉了dHDL抑制的自噬(圖6A–G),證明miR-181a-5p介導了HDL對自噬的調節。
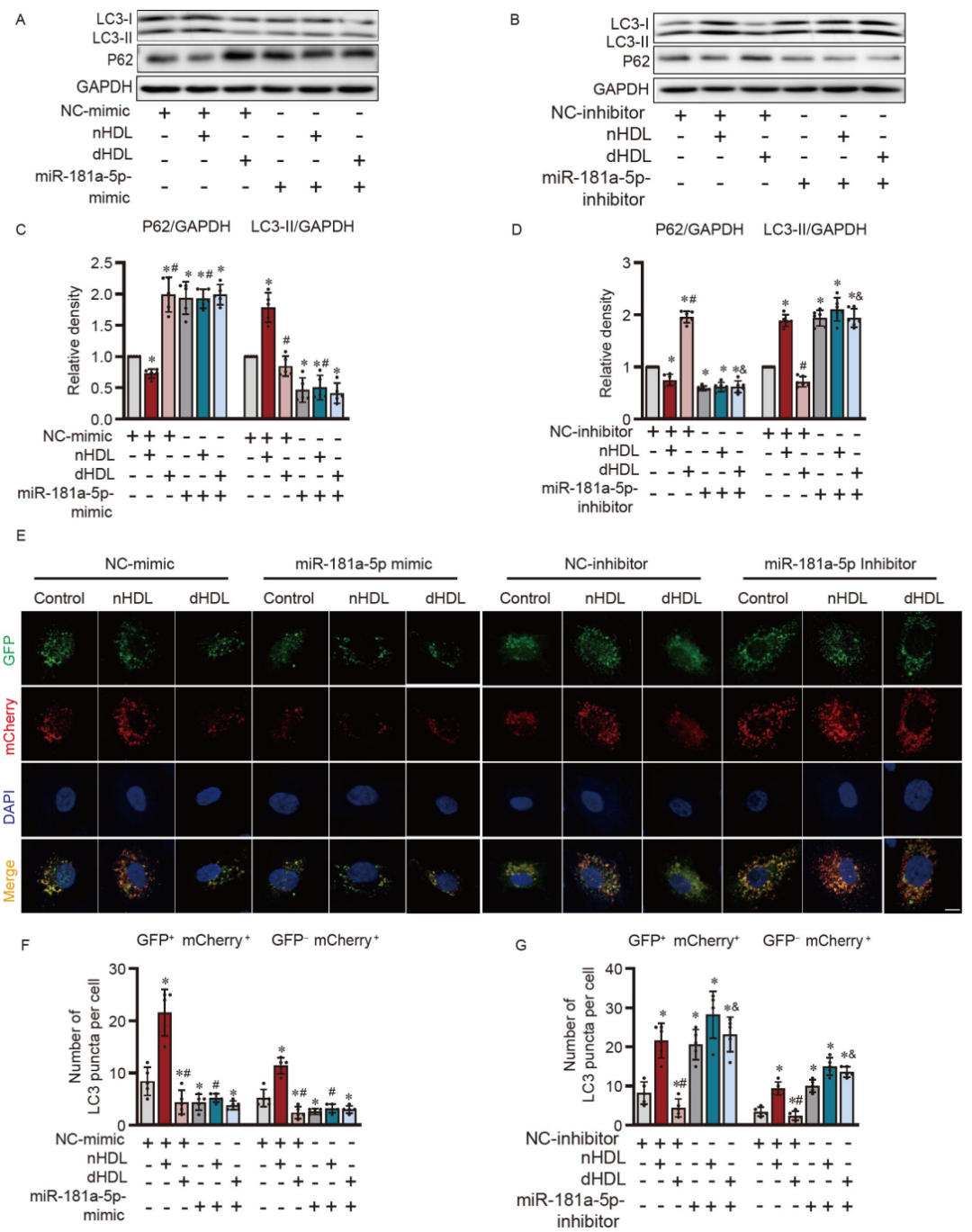
圖6. HDL對內皮自噬和miR-181a-5p調控的影響。
A和B,WB顯示轉染miR-181a-5p模擬物或抑制物的HUVECs在nHDL或dHDL刺激后LC3-I/II和P62的表達;C和D,A和B中圖像的定量分析;E,共聚焦圖像顯示在轉染miR-181a-5p模擬物或抑制物后,通過在HUVECs中使用自噬通路報告基因mCherry-GFP-LC3來觀察nHDL和dHDL對自噬的影響;F和G,合并圖像中測量的自噬體(GFP+mCherry+)和自溶體(GFP−mCherry+)的定量。
nHDL和dHDL通過miR-181a-5p不同程度地影響內皮細胞的NO產生、O2•−生成、遷移和血管形成
研究者進一步研究了HDL是否能通過miR-181a-5p來調節內皮功能。圖7A–D顯示,miR-181a-5p模擬物抑制了nHDL誘導的內皮細胞中的NO產生,而miR-181a-5p抑制劑逆轉了dHDL抑制的NO產生。miR-181a-5p模擬物增加了nHDL處理的內皮細胞中的O2•−生成(圖7E和G),而miR-181a-5p抑制劑降低了dHDL誘導的O2•−生成(圖7F和H)。此外,miR-181a-5p模擬物顯著抑制了nHDL誘導的內皮細胞遷移(圖8A和E)和血管形成(圖8C和G)。miR-181a-5p抑制劑恢復了dHDL抑制的內皮細胞遷移(圖8B和F)和血管形成(圖8D和H)。這說明nHDL通過抑制miR-181a-5p來激活自噬,從而誘導NO產生和血管生成,而dHDL則通過增加miR-181a-5p并抑制自噬,從而減少NO產生并刺激O2•−生成,以抑制血管生成。
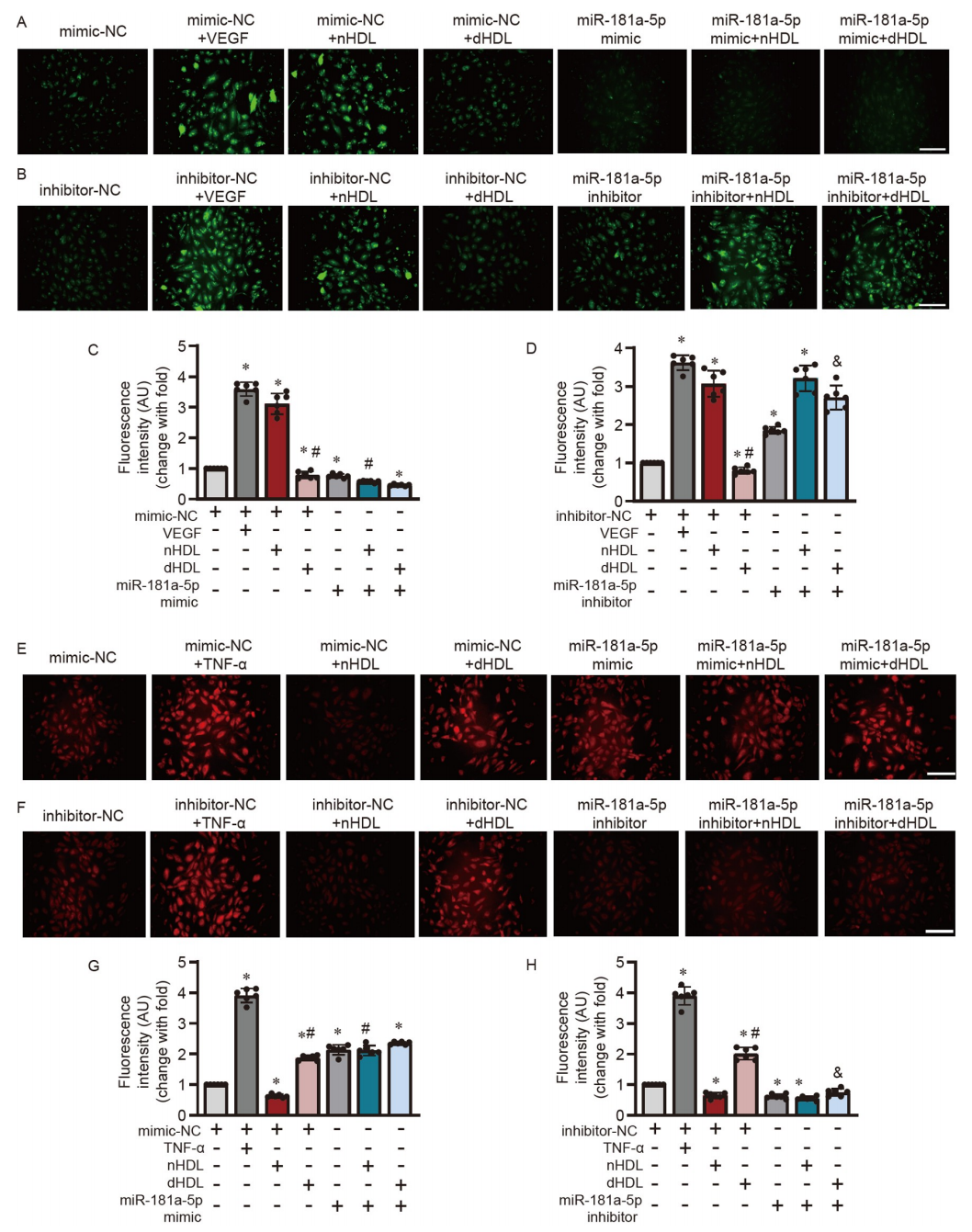
圖7. HDL通過調節miR-181a-5p影響HUVECs內皮細胞的NO產生和O2•−生成
A和B,免疫熒光分析顯示miR-181a-5p模擬物或抑制劑轉染后,HUVECs在nHDL或dHDL刺激下的NO產生;C和D,對A和B中圖像的定量分析;E和F,顯示miR-181a-5p模擬物或抑制劑轉染后,HUVECs在nHDL或dHDL刺激下的O2•−生成;G和H,對E和F中圖像的定量分析。
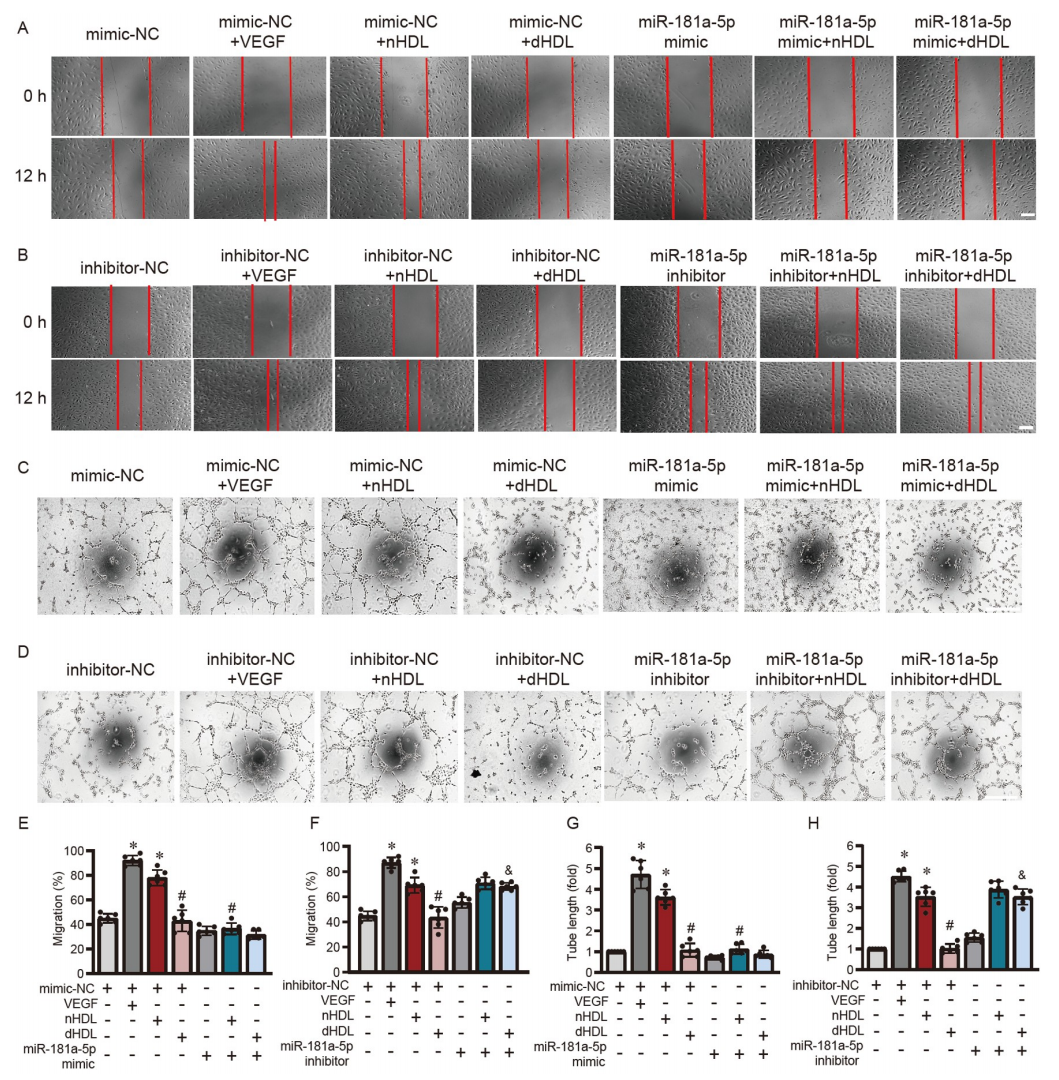
圖8. 通過調節miR-181a-5p/ATG5/eNOS軸調控HDL對內皮功能的影響
A和B,顯示miR-181a-5p模擬物或抑制劑轉染后,HUVECs在nHDL或dHDL刺激下的遷移;C和D,顯示miR-181a-5p模擬物或抑制劑轉染后,HUVECs在nHDL或dHDL刺激下的血管形成。
ATG5的過表達促進了高膽固醇LDLr−/−小鼠的血管生長
在高膽固醇低密度脂蛋白受體缺失(LDLr−/−)小鼠的HDL中,脂質過氧化物水平明顯高于C57BL/6小鼠,表明LDLr−/−小鼠中的HDL具有促炎性。因此,使用高膽固醇LDLr−/−小鼠的缺血下肢來確定自噬對體內血管生長的影響。與C57BL/6小鼠相比,高膽固醇LDLr−/−小鼠的缺血下肢中ATG5和eNOS的表達降低(圖9A和B)。過表達ATG5以增加自噬促進了eNOS的表達(圖9A和B),改善了缺血下肢的血流(圖9C和D)以及側枝血流(圖9E和F)。這些數據提供了在高膽固醇血癥中增強自噬可以促進血管生成的體內證據。
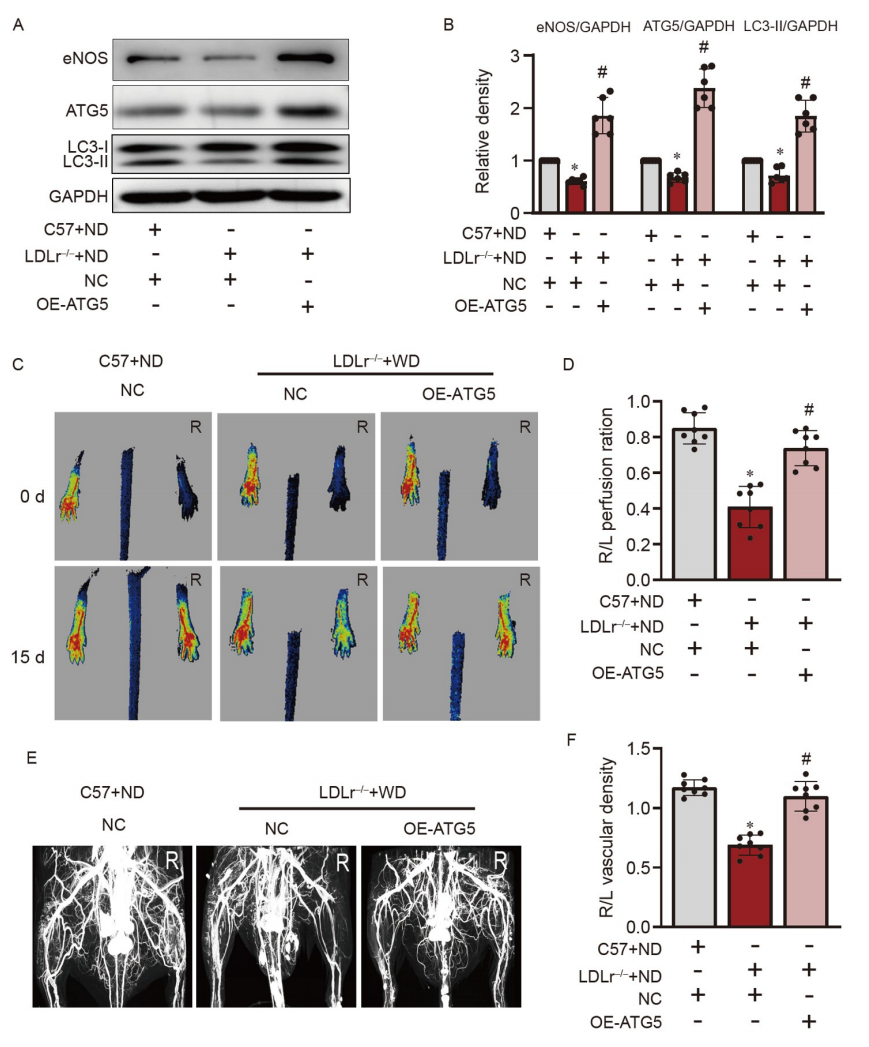
Figure 9. 通過上調ATG5促進體內血管生長
A和B,WB分析顯示LDL受體敲除(LDLr−/−)小鼠缺血下肢的脛骨肌中LC3-II、ATG5和eNOS的表達;C和D,血管生成分析的代表性圖像和定量分析;E和F,側枝分析的代表性圖像和定量分析。
總結
本研究揭示了nHDL通過調節內皮自噬促進血管生成,而dHDL通過增加miR-181a-5p來抑制內皮自噬,從而減少血管生成。ATG5的過表達能夠增加缺血下肢的血流和側支循環,從而促進血管生成。這項研究首次提供了nHDL通過調節自噬促進血管生成的直接證據,同時也為dHDL通過影響自噬抑制血管生成提供了新的認識。
參考文獻:
Bi-Ang Kang, Zhi-Jun Ou, Jing-Song Ou, et al. High-density lipoprotein regulates angiogenesis by affecting autophagy via miRNA-181a-5p. SCIENCE CHINA Life Sciences, 2023.
最新動態
-
09.23
中藥的現代詮釋:外泌體如何革新傳統醫學?
-
07.02
1+1>2!深度解析RNA測序數據挖掘邏輯和后期實驗設計思路,輕松研獲10+ SCI
-
07.01
“稻”亦有道——盤點近期水稻研究的重大突破
-
06.28
科學與美學的結合體:植物亞細胞定位技術詳解
-
06.28
“聚焦新質生產力,激發科研新動能”|LCA躋身蛋白互作研究的新銳力量
-
06.05
知無不“研”|一文讀懂免疫共沉淀技術(Co-IP)
-
05.14
四大研究利器(Co-IP、BIFC、Y2H、GST pull-down)助力速配蛋白互作“最佳拍檔”
-
05.14
高效、精準、直觀、實時——取經“蛋白互作研究翹楚”BIFC!
-
05.14
轉染效率低、干擾效果差、重復性欠佳...siRNA研究頻遇“攔路虎”怎么辦?
-
04.22
一文讀懂EMSA技術核心要點,讓“emsa” 秒變“easy”
 X
X 






