
SWATH-MS的那些事
信息來源:金開瑞 作者:genecreate 發布時間:2018-11-27 15:01:22
SWATH-MS基本概念
SWATH-MS檢測通常依賴于質譜的一級快速掃描 (通常至少10 Hz),高分辨(通常MS2的FWHM分辨率至少15000)以及高質量精度 (至少50ppm) ,其常見配置為第一級質量分析器為四級桿 (Q)而二級質量分析器為飛行時間(TOF)或Orbitrap(圖3A)。近年來在四級桿設計上的技術進步使其能夠有效的在整個近乎方形的母離子隔離窗口中傳輸離子(圖4B)。這個重要的改進保證了獲取到的信號的強度,并因此被檢測肽段的定量值不再會因為位于隔離窗口的不同位置而變化了。
在SWATH-MS數據采集過程中,質譜反復記錄了整個色譜梯度時間內一系列MS2掃描信息。而每個采集循環中MS1掃描信息可以只記錄一次或多次 (圖3A)。 其中關鍵的一點就是所有的MS2掃描是如何進行的與這些MS1并無關系。在每個MS2掃描時,都會隔離出一個相當寬的母離子窗口。該母離子隔離窗口的m/z會逐步遞增,直到完整掃描預設的整個質量范圍(圖3A)。為了保證足夠快速的記錄MS1和MS2以進行色譜峰提取和肽段定量,需要對MS2掃描數量,每個掃描的累積時間,母離子隔離窗口大小,肽段掃描質量范圍等參數均需要進行優化權衡。比如,最早提出SWATH-MS的Gillet用的采集方案是32個相同的25m/z的母離子隔離窗口,覆蓋了400到1200m/z的質量范圍,每個掃描的累積時間是100ms (圖3B)。
緊接著而來的一個重要結論就是如此大的母離子隔離窗口將會同時引入大量肽段,并將它們同時碎裂(圖3C和1D)。取決于樣品復雜度,這些窗口里會同時引入數十個甚至數百個共碎裂的肽段(Rost等,2012),從而產生了高度混合而卷積的MS2譜圖。如此高度多路的MS2譜圖無法再用傳統的基于數據庫匹配的DDA譜圖搜索軟件直接進行分析。因此SWATH-MS數據需要更先進的數據處理策略。當然,DDA和靶向質譜通常也要設置0.5到3個m/z的隔離窗口,使得其碎片譜圖一樣會有來自于多個母離子的卷積碎片譜峰,但畢竟這樣的譜峰復雜度還是遠遠低于DIA的(Michalski等,2011; Wang等,2014)。解決方法之一稱之為“peptide-centric scoring” (Ting等,2015)。在這項策略中,采集到的數據集會和一套已知肽段各類參數的數據集進行比對搜索。最常用的方法就是預先采集一套譜圖數據庫。該策略也被稱之“targeted data extraction”,也是在最早發表的SWATH-MS文獻中提出 (Gillet等,2012) ,而至今為止也是分析DIA數據的主要手段。總的來說,在SWATH-MS實驗中,所有落在隔離窗口范圍內的離子化的多肽都將被同時碎裂并將被系統化的、無歧視的一并記錄下來。SWATH-MS數據的定性和定量分析依賴于從高度多路的MS2譜圖中提取,因此非常依賴于先進的數據處理策略,比如peptide-centric scoring。SWATH-MS是對追求蛋白質組全覆蓋的DDA技術和追求高可重現性的靶向蛋白質組的一種補充。
SWATH-MS的優勢
從上面總結的SWATH-MS的原理可以推導出其可能的性能優勢和限制,我們在表1中進行了總結。SWATH-MS 和其他DIA技術的優勢場景及其近期流行的原因均在引文中總結(Doerr,2015) ,接下來,我們也對其優勢進行討論。
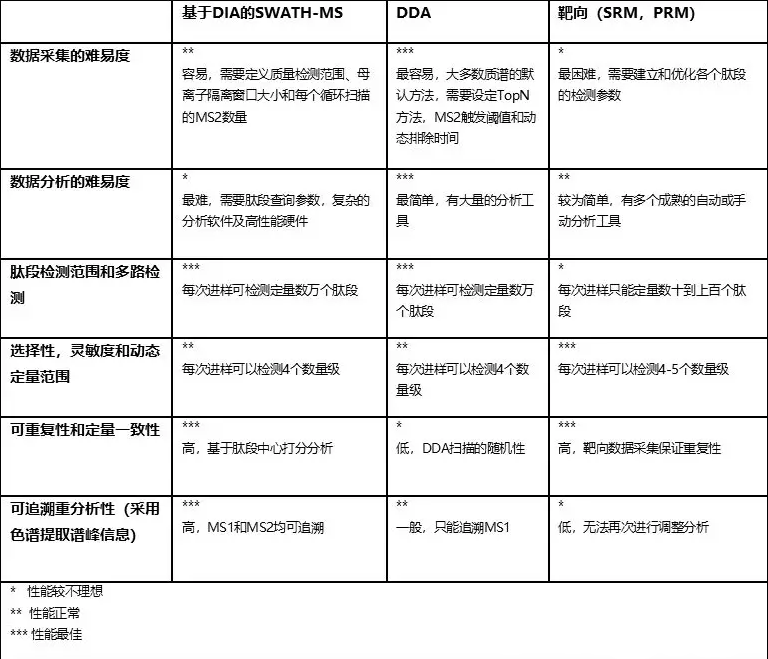
表1 優勢和劣勢:SWATH-MS和DDA及靶向(SRM,PRM)蛋白質組技術比較
1.數據采集的難易度
SWATH-MS的一項重要技術優勢就是簡化了數據采集流程。一次SWATH-MS采集需要定義母離子隔離方式(包括窗口峰寬、m/z采集范圍,MS2掃描累積時間)。一旦方法確認,同一套系統中所有相似類型的樣品均可采用統一的相同方法進行實驗分析了。即使樣品類型不同,同樣的采集方法也被證明依然很可靠。這種數據采集方式的簡便性和DDA采集差別不大,比靶向方法來得簡單得多。像peptide precursor ions (PRM)或者peptide-fragment ion pairs (SRM)就需要預先定義每個感興趣肽段的檢測參數,且采集時經常需要確認好相應的保留時間 (Picotti & Aebersold,2012)。
2.肽段檢測范圍和多路檢測
SWATH-MS的第二項優勢是其對肽段定性和定量的高多路性能(一張MS2譜圖含有多個肽段的碎片離子)。和基于DDA的方法類似,不同于靶向蛋白質組學,單次SWATH-MS檢測可以定性超過10000種肽段。這早已在諸如人細胞系HEK293中進行了驗證,通常這類樣品在1%蛋白FDR水平下,2小時檢測可以找30000到40000種多肽,對應于4000-5000種蛋白質 (Collins等,2017)。因此,相對于SRM或PRM方法,SWATH-MS實現了巨大的定量肽段數量提升。定量肽段越多,相應的可定量蛋白也越多,同時檢測到的蛋白序列覆蓋度也越高。這樣可以改善蛋白組裝的效果,相應帶來更精確和準確的蛋白定量結果,進一步也可以幫助進行蛋白質翻譯后修飾和可變剪切的檢測。還有就是高多路檢測率也可以提升全局數據歸一化的效果。我們通常認為各個樣品間大部分蛋白的濃度應該是不變的,但在SRM和PRM中,通常只檢測目標肽段,因此按照這個邏輯進行樣品間的全局歸一化非常困難(尤其是目標肽段通常都是我們假定是參與的研究中生物學過程調控的蛋白)。SWATH-MS和DDA一樣可以平行檢測一系列在生物調控過程中無偏差的肽段,這是全局歸一化的前提條件。
3.可重復性和定量一致性
當研究數百的樣品的蛋白質組學時,肽段檢測的可重復性及定量一致性就變成了主要困難。在DDA蛋白質組實驗中要求肽段定量的高度一致性是非常困難的,因為你無法要求質譜DDA實時采集信號的時候所用的啟發式母離子選擇方式能夠實現可重復性。因此當一個肽段在某次DDA檢測中沒有被定性成功,你不能判斷該肽段到底是否可檢出(“真陰性肽段定量”)。其實際情況很可能僅僅是因為質譜恰好因為一堆共流出肽段的存在,沒有去碎裂這條多肽(“假陰性肽段定量”) (Michalski等,2011)。當然也有相應的降低這些假陰性的改進方法,比如嘗試將某些樣品中定性成功的肽段信息在不同樣品中進行傳遞,從而大大改善定量數據矩陣的完整性 (Cox等,2014; Mueller等,2007; Prakash等,2006)。
相反的,在SRM,PRM和SWATH-MS中,MS2數據采集相當的有系統性且分析策略是以肽段為中心。通過這些方法的結合,我們可以較為可信的推導出目標肽段是的確“沒被檢測到”并因此推斷出該樣本中此肽段的含量至少是低于最低檢測限的。盡管如此,當我們用自動化數據分析工具來檢索數千種蛋白或數萬種肽段時,SWATH-MS的 “假陰性肽段定量”在某種程度上依然存在。當然,和DDA一樣,SWATH-MS的分析軟件(Rost等,2016)也會進行樣品間譜峰間對齊和鑒定結果轉移。相對于母離子檢測來說,碎片離子檢測的靈敏度更高,且包含的信息也更豐富 (MS2中,肽段能夠共流出大量碎片離子而MS1中一個肽段只能流出一個離子),因此這種結果轉移方式相對于DDA來說更穩健、更靈敏。
最后一點,SRM (Abbatiello等,2015; Addona等,2009)和SWATH-MS (Collins等,2017)技術都被認為能夠實現非常好的跨實驗室和實驗室內的結果重現性,且變異系數能夠低于20% CV,即使在多個不同國家實驗室進行的大規模研究也能達到這種程度。因此,如此高的重復性和一致性可以幫助生物學研究解決很多問題 (Rost等,2015)。
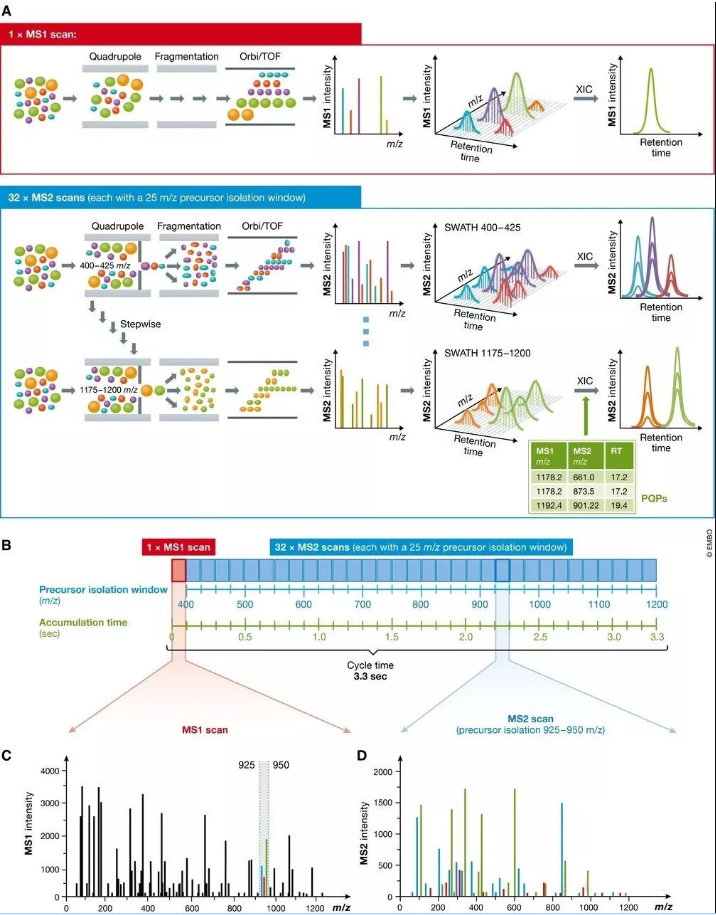
圖3 SWATH-MS技術中順序窗口數據非依賴采集的原理
(A) SWATH‐MS檢測通常在具備快速質量掃描的雜交質譜上進行,主要在第一級質量分析器是四級桿而第二級質量分析器是TOF或Orbitrap的質譜上進行。在SWATH‐MS模式下,通常MS1掃描一次后會緊跟著一系列母離子隔離窗口很寬的MS2譜圖(比如25 m/z的窗口大小)。通過在預先設定的質量范圍內反復循環的連續母離子隔離掃描,質譜能夠記錄相對完整的譜圖數據,其中包含了所有可檢測的母離子和碎片離子的完整而連續譜峰信息。由此,MS1和MS2的提取離子色譜均可由此產生。而SWATH‐MS數據的分析則可以采用基于肽段的打分策略,需要預先記錄所有可檢索的肽段的色譜及質譜特征信息,從而合成一個由peptide query parameters (PQPs)構成的知識庫來幫助進行肽段匹配。(B) 由Gillet et al (2012)提出的SWATH‐MS數據采集流程,在Q-TOF質譜上采集,采用預先設定的32個相同的25m/z的母離子隔離窗口,覆蓋了400到1200m/z的質量范圍,每個完整掃描的累積時間是100ms,完整的循環時間是3.3秒。 (C) 在某個時間點流出的MS1全掃描能夠檢測到的所有肽段母離子。比如質量范圍在925到950 m/z,有三個共流出肽段被檢測到(綠色,紅色和藍色標記)。 (D) 925–950 m/z范圍內這3個肽段母離子產生的混合碎片離子MS2譜峰
4.可追溯查詢
SWATH-MS數據的色譜和質譜的MS1母離子及MS2碎片離子譜峰信息都相當完整。這和DDA數據形成鮮明對比,DDA采集時只有母離子是連續記錄而碎片信息則并不完整。SWATH-MS數據因此非常適合于過了一段時間后的追溯性分析,在最初的生物學假設中并不關注的信息或者早期無法確認的一些肽段分析參數,比如新獲得的蛋白序列、肽段或者新的后修飾就可以再次進行檢索分析(Rosenberger等,2017)。這不像SRM或PRM分析,如果要檢測新的蛋白或肽段就必須得重新實驗。
5.分析修飾肽段
這個優勢是其對修飾肽段的定性和定量能力,它能夠定位肽段序列中發生修飾的氨基酸位點,也能夠搜索之前非預期的分析物。這些重要的能力是由SWATH-MS檢測中對肽段整個流程時間內循環記錄其高分辨、高精度MS2譜圖所帶來的收益。因此,不像DDA由于存在隨機采集MS2和動態排除等問題,SWATH-MS可以將數據中的XIC相關MS1和MS2的信息最大化保留。還有就是對于PTM位點定位來說,碎片離子決定了修飾氨基酸的位置并且完整的MS2譜圖可以用來判斷不同可能位點間置信度差異(Keller等,2016; Meyer等,2017; Rosenberger等,2017)。最后一點,SWATH-MS和開放式修飾搜索工具結合可以發現非預期的全新修飾,且并不會造成搜索空間的組合爆炸 (Keller等,2016; Wang等,2015)。然后,當采用SWATH-MS來分析修飾肽段時也有和其他鳥槍法蛋白質組檢測方式一樣的固有挑戰,亦即是修飾肽段大部分的濃度總是遠遠低于非修飾肽段。
總的來說,以上總結的SWATH-MS性能優勢可以對復雜樣本中海量的肽段進行精確定量,并能達到極高的重復性和一致性。典型的應用場景包括系統生物學研究 (Bensimon等,2012),基因相關的研究 (Liu等,2015; Okada等,2016; Williams等,2016) ,臨床普篩(Liu等,2014; Sajic等,2015),藥物/干擾組學普篩(Litichevskiy等,2017) 或者探索性的基礎研究 (Collins等,2013; Lambert等,2013; Parker等,2015; Schubert等,2015b; Selevsek等,2015)。SWATH-MS同樣適合進行快速檢測分析,并能夠單針檢測就實現在復雜哺乳動物樣本中約50%的蛋白質組覆蓋率(Bruderer等,2017)。
SWATH-MS的缺陷和挑戰
1.先驗知識和預構建譜圖庫的獲取難度
在最常規且成功應用SWATH-MS的分析流程中,肽段中心打分算法依賴于關于目標肽段的色譜、質譜特性先驗知識所形成的高質量譜圖庫 (Schubert等,2015a)。通常譜圖庫由DDA方式產生,由此帶來的問題就是只有DDA實驗中能夠檢測到的肽段才可能在DIA中分析到。最近幾年,不依賴于譜圖庫的SWATH-MS 數據分析替代方案也有了長足發展,比如DIA-Umpire (Tsou等,2015),FT-ARM (Weisbrod等,2012),和PECAN (Ting等,2017)。
2.數據分析的難度
目前,SWATH-MS 實驗分析中主要的挑戰就是肽段中心打分算法部分。高度多路的MS2譜圖可能由多達10個到100多個共碎裂的肽段母離子形成,因此依賴于一套精巧的分析路線來檢測和定量肽段并將它們進行可靠的統計分析。對于DDA蛋白質組學來說,20多年來已經發展了豐富的分析路線 (Cox & Mann,2008; Deutsch等,2015; Keller等,2005; Reinert & Kohlbacher,2010)。而靶向蛋白質組學分析軟件也已經成熟應用了好幾年了(Colangelo等,2013; Malmstrom等,2012; Teleman等,2012)。相應的自動化分析SWATH-MS肽段譜的軟件還處于發展階段,其中一些也已經得到了廣泛應用并供免費使用(相應的綜述參考 (Bilbao等,2015))。對其中五種廣泛使用的軟件的綜合比較文獻可以參考(Navarro等,2016)。
3.選擇性,靈敏度和動態定量范圍
SWATH-MS還有一個主要的挑戰是其定量的態范圍依然有限。多個課題組報道了在大規模復雜蛋白質組樣本中碎片離子(MS2)具有比相應母離子(MS1)更高的信噪比,選擇性和同一個掃描內的動態范圍(Egertson等,2013; Gillet等,2012; Venable等,2004)。在復雜樣本中,肽段的MS1信號很有可能被共流出的非常接近或相同m/z的信號所干擾。而在MS2水平的信號則僅來自于MS1隔離窗口內的這些母離子信號碎片,而由于MS2定量會選擇多個離子碎片,其均受到干擾的可能性非常之低。另外考慮到離子阱質譜的選擇性能力,MS1掃描的信號受到其AGC(auto gain control)效應的影響,高豐度肽段信號會抑制低豐度肽段,而SWATH-MS中由于只檢測隔離窗口內的母離子碎片,該效應得到很好地改善。目前SWATH-MS在Q-TOF儀器上使用時,可以在單針檢測復雜的注入人細胞系全酶解液的實驗中覆蓋4到4.5個數量級定量動態范圍(Collins等,2017)。文獻報道中的復雜樣品定量最低檢測限大約為中等attomole到低femtomole(上柱量)。如此定量范圍還是比最新的SRM或MRM檢測的靈敏度低3-10倍 (Gillet等,2012; Liu等,2013; Schmidlin等,2016) 。
綜上,目前相對于SRM或PRM技術來說,SWATH-MS的主要劣勢在于其靈敏度較低。因此,對于尤其關注低豐度肽段精確定量的項目來說,最佳的選擇方案就選擇一部分最感興趣的目標肽段進行靶向定量。另外,大家也要注意SWATH-MS檢測需要在譜圖庫和肽段查詢流程建立和優化上花費較多的功夫。
最新動態
-
09.23
中藥的現代詮釋:外泌體如何革新傳統醫學?
-
07.02
1+1>2!深度解析RNA測序數據挖掘邏輯和后期實驗設計思路,輕松研獲10+ SCI
-
07.01
“稻”亦有道——盤點近期水稻研究的重大突破
-
06.28
科學與美學的結合體:植物亞細胞定位技術詳解
-
06.28
“聚焦新質生產力,激發科研新動能”|LCA躋身蛋白互作研究的新銳力量
-
06.05
知無不“研”|一文讀懂免疫共沉淀技術(Co-IP)
-
05.14
四大研究利器(Co-IP、BIFC、Y2H、GST pull-down)助力速配蛋白互作“最佳拍檔”
-
05.14
高效、精準、直觀、實時——取經“蛋白互作研究翹楚”BIFC!
-
05.14
轉染效率低、干擾效果差、重復性欠佳...siRNA研究頻遇“攔路虎”怎么辦?
-
04.22
一文讀懂EMSA技術核心要點,讓“emsa” 秒變“easy”
 X
X 






